
 商标分类
商标分类  商标转让
商标转让 
桃氰丙氨酸合成酶基因在提高植物耐盐性方面的应用的制作方法
 2021-02-02 18:02:50|
2021-02-02 18:02:50| 401|
401| 起点商标网
起点商标网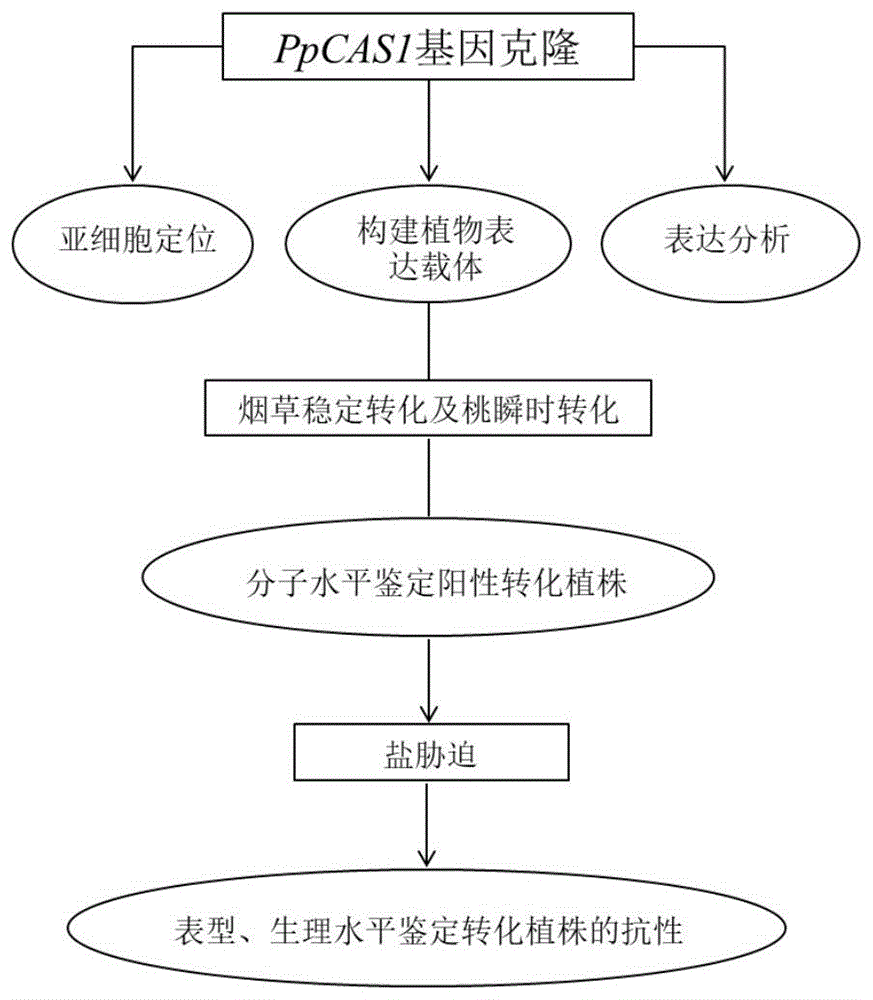
本发明属于桃耐盐相关基因领域,特别涉及桃氰丙氨酸合成酶基因在提高植物耐盐性方面的应用。
背景技术:
桃[prunuspersica(l.)batsch],蔷薇科,原产于我国西北地区,是我国栽培历史悠久分布区域广泛的主要果树之一。近年来我国桃树栽培面积稳步发展,桃产量和消费量均处于世界领先地位(usda2017)。桃树根系在土壤中分布较浅,且需氧量较高,植株的生长发育很容易受到各种环境因素的影响,如盐碱,干旱,涝害等。因此,发掘抗逆基因资源,创制抗性强的种质资源和培育优良的抗逆新品种,成为了桃产业健康可持续发展的重要因素。
土壤盐渍化是当今世界农业生产中最主要的环境限制因子之一,严重影响植物的种子萌发、作物生长发育和产量。高浓度盐碱环境对植物体细胞内的离子平衡产生破坏,使植物呈现离子中毒症状,同时会产生严重的渗透胁迫,氧化胁迫,影响植物细胞的吸水能力,降低植株光合效率。最后,盐胁迫逆境产生的负面影响会导致植物体整个生理及代谢过程发生紊乱,严重时将造成植物死亡(zhu2002)。植物在漫长的生物进化过程中逐渐演变出一系列复杂的响应机制来应对环境中的盐胁迫,主要包括离子平衡的重建、细胞内渗透水平的调节及活性氧的清除等(zhu2001;yangandguo2018)。
β-氰丙氨酸合成酶(β-cyanoalaninesynthase,cas)是高等植物氰化物同化途径中的关键酶,氰化物在β-氰丙氨酸合成酶作用下与半胱氨酸结合形成β-氰丙氨酸,同时释放硫化氢,接着β-氰丙氨酸在兼具腈水解酶(nitrilase)和腈水合酶(nitrilehydratase)的nit4作用下产生天冬酰胺或天冬氨酸和氨气(garcíaetal.2010)。氰化物经同化途径最终产生氨基酸可供植物生长发育(machinguraetal.2016)。前人研究表明:β-氰丙氨酸合成酶在植物应对非生物胁迫中发挥重要的作用,如liang发现烟草在干旱胁迫下,利用同工酶技术发现β-氰丙氨酸合成酶活性显著提高(liang2003)。拟南芥β-氰丙氨酸合成酶缺失突变体对水分亏缺更加敏感(machinguraetal.2013)。yu等超表达烟草中β-氰丙氨酸合成酶基因,发现可以提高植株的耐盐性(yuetal.2020)。桃属于典型的多年生木本生氰植物,与模式植物拟南芥、烟草等亲缘关系较远。目前,在桃中还未曾有过β-氰丙氨酸合成酶基因的相关报道。因此,深入研究桃中β-氰丙氨酸合成酶基因在抗逆方面的功能具有重要的意义。
技术实现要素:
本发明的所要解决的技术问题是提供桃氰丙氨酸合成酶基因在提高植物耐盐性方面的应用。
本发明解决上述技术问题的技术方案如下:
桃氰丙氨酸合成酶基因在提高植物耐盐性方面的应用,所述桃氰丙氨酸合成酶基因的序列为seqidno:1。
本发明的有益效果为:本发明发现过表达桃氰丙氨酸合成酶基因(ppcas1)能够有效的增强转基因植株的活性氧清除能力,从而提高了植株的耐盐性,可应用于桃种植领域,提高种植收益,降低种植成本。
附图说明
图1是本发明的方法流程示意图;
图2是桃β-氰丙氨酸合成酶基因ppcas1与梅,苹果,拟南芥等植物氨基酸序列对比结果构建的进化树图;
图3是本发明实施例2中ppcas1基因在盐,脱水,低温和aba处理下的表达示意图;
图4是本发明实施例3中ppcas1蛋白亚细胞定位结果图;
图5是本发明实施例4中ppcas1转基因烟草植株的获得流程图;
图6是本发明实施例5转ppcas1的编码基因株系及野生型(wt)氯化钠处理前后的表型和生理指标测定图;
图7是本发明实施例5中的ppcas1沉默植株(ptrv-1,ptrv-2)及对照植株(wtp)盐处理前后表型观察及相关生理指标测定结果;
图8是本发明实施例5提供的转ppcas1的编码基因烟草和利用病毒介导的vigs技术瞬时沉默ppcas1桃组织化学染色分析h2o2和o2-积累结果图,;
图9是本发明实施例5提供的转ppcas1的编码基因烟草和利用病毒介导的vigs技术瞬时沉默ppcas1桃中抗氧化酶活性测定结果图。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
以下通过实验对桃ppcas1基因提高桃植株耐盐性的效果进行验证。一、桃ppcas1序列分析及基因全长cdna的克隆
1、ppcas1基因序列分析
用拟南芥中已经通过功能验证的β-氰丙氨酸合成酶的蛋白序列,通过蛋白序列blast在桃基因组中找到同源性最高的序列,具体氨基酸序列如下所示:
maalrsflkkrstllcneavrkrlfstqvsqpiidspsfaqrvrnlpkdlpgthvktevsqligrtpivylnkvtegcgayiavkqemfqptssikdrpalsmindaekkglitpgktvlveptsgnmgismafmaamrgykmvltmpsytslerrvcmrafgaeliltdpargmggtvkkaydllestpnahmlqqfsnpantrvhfettgpeiwedtdgqvdifimgigsggtvsgvgqylksknpnvqiygvepaesnvlnggkpgphlitgngvgfkpdildldvmdrvievksddavkmarrlaleeglmvgissgantvaaielakkpenkgklivtvhpsfgerylssvlfeelrkeaenmqpvsvd
具体核苷酸序列如下所示:
atggcggccttgaggagctttctgaagaaaagatccactttgctctgcaatgaagccgtgagaaagagactcttctcaacccaagtgagccaacccatcattgattctccatcttttgctcagagggtcagaaaccttcctaaggatcttcctggaacccacgtcaaaacagaagtctcccaactcattggtagaactcccattgtctatctcaacaaagtcactgaaggatgtggagcttatatagctgtcaagcaagagatgtttcaacccacctctagcatcaaagacagaccagcactttcaatgatcaacgatgcagaaaagaaaggcttgataactcctggcaagacagtactggtggagccaacatcaggaaatatgggaatcagcatggcttttatggcagccatgagagggtataaaatggttctcactatgccgtcttacacaagcttggagagaagggtgtgtatgagagcctttggagctgaattaattctcactgatccagccagggggatgggaggaactgttaagaaggcttatgatcttctggaatccacaccaaatgctcatatgctccaacagttctcaaatcctgccaatactcgggtgcatttcgaaactacaggccctgagatatgggaggataccgatgggcaagttgacatcttcataatgggaattggtagcggaggcacagtctctggggttgggcagtatctcaaatccaaaaatcccaatgttcagatatatggagtggagcctgctgaaagtaatgtgctaaacggtggcaaaccaggtcctcatttgatcactggcaacggggttggattcaaaccagatattttggacttggatgtgatggatagagttattgaggttaaaagtgatgatgcagtaaaaatggcaagacgattggcattggaagagggacttatggtaggaatatcatccggagccaacacggtggcagcaattgagcttgctaaaaagccagaaaacaaaggcaaacttattgtgaccgttcacccaagtttcggggagcgatacttgtcatctgtcctgtttgaagagctgaggaaggaagctgaaaacatgcaaccggtctcagtcgactaa
生物信息学分析cdna序列显示,ppcas1基因全长1125bp,它包括的编码阅读框编码374个氨基酸,等电点为8.78,预测的分子量为40.69kda。expasy分析表明编码的氨基酸ppcas1有线粒体定位信号。根据氨基酸的多序列对比结果构建的进化树来看,推导的ppcas1的氨基酸序列与梅pmcas1、甜樱桃pacas1、苹果mdcas1和mdcas2序列高度同源。根据氨基酸的多序列比对构建的进化树来看,桃ppcas1蛋白同梅pmcas1亲缘关系最近,甜樱桃pacas1、苹果mdcas1和mdcas2次之,同玉米zmcys和高粱sbcas2进化上离得最远(图2)。
2、ppcas1基因克隆
2.1rna提取
研究材料桃实生苗种植在华中农业大学果树基地,其苗龄为30天。挑选生长健壮,长势一致的30株幼苗混合,迅速用液氮进行速冻。rna提取采用北京艾德莱生物科技有限公司easyspinplusplantrnakiteasyspinplus植物rna快速提取试剂盒。具体方法如下:
(1)取500μl裂解液rlt,转入1.5ml离心管中,加入50μlplantaid混匀备用。
(2)液氮中研磨适量植物组织成细粉后,取50-100mg细粉转入上述装有rlt和plantaid的离心管,立即用手剧烈振荡20s,充分裂解。
(3)将裂解物13000rpm离心10min,取裂解物上清转到一个新离心管。加入上清体积一半的无水乙醇,此时可能出现沉淀,但是不影响提取过程,立即吹打混匀,不要离心。
(4)将混合物(每次小于720μl,多可以分两次加入)加入一个基因组清除柱中,(清除柱放入收集管中)13000rpm离心2min,弃掉废液。
(5)将基因组dna清除柱子放在一个干净2ml离心管内,在基因组清除柱内加500μl裂解液rltplus,13000rpm离心30s,收集滤液(rna在滤液中),用微量移液器较精确估计滤过液体积(通常为450-500μl左右,滤过时候损失体积应该减去),加入0.5倍体积的无水乙醇,此时可能出现沉淀,但是不影响提取过程,立即吹打混匀,不要离心。
(6)立刻将混合物(每次小于720μl,多可以分两次加入)加入一个吸附柱ra中,(吸附柱放入收集管中)13000rpm离心2min,弃掉废液。
(7)加700μl去蛋白液rw1,室温放置1min,13000rpm离心30s,弃掉废液。
(8)加入500μl漂洗液rw(请先检查是否已加入无水乙醇),13000rpm离心30s,弃掉废液。加入500μl漂洗液rw,重复一遍。
(9)将吸附柱ra放回空收集管中,13000rpm离心2min,尽量除去漂洗液,以免漂洗液中残留乙醇抑制下游反应。
(10)取出吸附柱ra,放入一个rnasefree离心管中,加50μlrnasefreewater(事先在70-90℃水浴中加热可提高产量),室温放置1min,12000rpm离心1min。
将提取到的桃rna立即放至-80℃超低温冰箱中保存以备用。取2μlrna样品用于1%琼脂糖凝胶电泳,用nanodrop2000仪器(thermoscientific,美国)进行检测,检测其浓度为250ng/μl。
2.2基因克隆
运用primer5.0设计引物,ppcas1基因克隆引物如下:
正向引物:5’-atggcggccttgaggagct-3’
反正引物:5’-ttagtcgactgagaccggttgc-3’
取桃rna1μg参照primescriptrtreagentkitwithgdnaeraser(takara,中国)反转录试剂盒说明书进行操作,以得到的cdna为模板,采用南京诺唯赞公司高保真酶进行ppcas1基因扩增。反应体系为:2×phantamaxbuffer25μl,dntpmix1μl,正向引物2μl,反向引物2μl,phantamaxsuper-fidelitydnapolymerase1μl,模板1μl,ddh2o18μl。
pcr按以下程序完成:
扩增完成后产生单一条带的pcr产物,经1%的琼脂糖凝胶电泳后,用生工生物工程股份有限公司胶回收试剂盒按照使用说明,回收特异目的条带。回收纯化的溶液与北京全式金生物技术有限公司peasy-bluntzero载体进行连接,连接体系中基因与载体的摩尔比为7:1连接。反应总体积是5μl,其中1μl载体,4μl的pcr纯化的产物。25℃连接5分钟后,采用热击法转化到大肠杆菌感受态trans5α中。
大肠杆菌转化方法如下:
(1)将5μl连接产物加入50μl冰浴融化的大肠杆菌感受态trans5α(全式金,中国)细胞中,轻轻混匀,冰上放置30min;
(2)42℃水浴中热激45s后,立刻转移至冰中放置2min,该过程不要摇动离心管;
(3)加入500μl不含抗生素的lb液体培养基,37℃摇床,200rpm培养1h,使细菌复苏;
(4)取100-200μl复苏后的感受态细胞均匀涂于含有相应抗生素的lb固体培养基,培养皿倒置放于37℃恒温培养箱中,培养过夜。
转化后12-16h挑取平板上单克隆于2ml离心管,加入含相应抗生素的lb液体培养基,37℃摇床震荡培养至菌液浑浊,然后吸取1μl菌液作为模板后进行阳性鉴定。试剂采用康为世纪2×taqmastermix(dye),反应体系为:2×taqmastermix(dye)12.5μl,正向引物1μl,反向引物1μl,模板1μl,ddh2o9.5μl。
阳性鉴定引物序列如下:
正向引物:5’-tttcatttggagaggactcc-3’
反正引物:5’-taacgtgactcccttaattc-3’
pcr程序为:
获得阳性克隆后,将阳性克隆进行测序(由武汉擎科生物科技公司完成)。测序正确的克隆摇菌并使用北京全式金生物技术有限公司质粒小提试剂盒提取菌液质粒
二、不同逆境处理下ppcas1基因的表达分析
为了分析桃中ppcas1基因对高盐,脱水,低温和aba处理的响应模式,使用实时荧光定量pcr(qrt-pcr)技术对ppcas1基因的表达模式进行分析。采用艾德莱试剂盒提取rna,cdna的合成参照primescriptrtreagentkitwithgdnaeraser(takara,中国)反转录试剂盒的操作手册进行。qrt-pcr反应使用上海翊圣生物科技公司hiefftmqpcrsybrgreenmastermix。qrt-pcr反应体系中有:2×mix5μl,cdna0.6μl,正向引物0.2μl,反向引物0.2μl,ddh2o4μl。
qrt-pcr反应程序为:
qrt-pcr引物序列如下
ppcas1-qpcr:
正向引物:5’-cttcctggaacccacgtcaa-3’
反向引物:5’-tccaccagtactgtcttgcc-3’
内参引物pptef2:
正向引物:5’-agcaagtcacccaacaagcata-3’
反向引物:5’-ccaaccaaactcttcagccaat-3’
qrt-pcr结果图3。图3为ppcas1基因在盐处理,脱水,低温和aba处理下的表达示意图。其中,图3a是所述编码基因在桃实生苗在100mmnacl处理下,相应时间点取样,采用qrt-pcr分析的相对表达量;图3b是桃实生苗在脱水处理下,处理后不同时间点的基因表达模式;图3c是桃实生苗在4℃处理下,相应时间点取样,采用qrt-pcr分析的所述编码基因的相对表达量。图3d是桃实生苗在100μmaba处理后不同时间点的基因表达模式。由图中可以看出,在100mmnacl处理下,ppcas1基因的表达量显著上升,在24小时达到最高点是最初的7.5倍。这表明ppcas1受盐胁迫的影响,是一个高盐响应的候选基因。
三、ppcas1蛋白的亚细胞定位
3.1亚细胞定位载体构建
利用gateway方法经过两轮pcr反应构建pearleygate101-ppcas1亚细胞定位载体,以前期获得的带有ppcas1全长片段的peasy-bluntzero载体质粒为模板进行第一轮pcr反应。反应引物序列如下:
正向引物:5’-aaaaagcaggctccatggcggccttgaggagct-3’
反正引物:5’-agaaagctgggttgtcgactgagaccggttgc-3’
反应体系为:2×phantamaxbuffer12.5μl,dntpmix0.5μl,正向引物1μl,反向引物1μl,phantamaxsuper-fidelitydnapolymerase0.5μl,模板2μl,ddh2o7.5μl。
pcr按以下程序完成:
扩增完成后取5μl反应产物经1%的琼脂糖凝胶电泳,若条带单一清晰,取1μl反应产物为模板进行第二轮pcr反应。反应引物序列如下:
正向引物:5’-ggggacaagtttgtacaaaaaagcaggct-3’
反正引物:5’-ggggaccactttgtacaagaaagctgggt-3’
反应体系为:2×phantamaxbuffer25μl,dntpmix1μl,正向引物2μl,反向引物2μl,phantamaxsuper-fidelitydnapolymerase1μl,模板1μl,ddh2o18μl。
pcr按以下程序完成:
扩增完成后产生单一条带的pcr产物,经1%的琼脂糖凝胶电泳后,用生工生物工程股份有限公司胶回收试剂盒按照使用说明,回收特异目的条带。
利用gatewaybpclonaseiienzymemix(invitrogen,美国),取上述胶回收产物1μl作为模板进行bp反应,反应体系为:模板1μl,pdonr2070.4μl,tebuffer(ph=8.0)1μl,bpenzyme0.6μl。25℃反应16h后,冰上静置5min,产物采用热击法转化到大肠杆菌感受态trans5α中(具体操作方法同上)。
pcr阳性鉴定方法同上,鉴定引物序列为:
正向引物:5’-ggggacaagtttgtacaaaaaagcaggct-3’
反正引物:5’-ggggaccactttgtacaagaaagctgggt-3’
获得阳性克隆后,将阳性克隆进行测序(由武汉擎科生物科技公司完成)。测序正确的克隆摇菌并使用北京全式金生物技术有限公司质粒小提试剂盒提取菌液质粒。以此质粒为模板利用gatewaylrclonaseiienzymemix(invitrogen,美国)进行lr反应,反应体系为:模板1μl,pearleygate1010.4μl,tebuffer(ph=8.0)1μl,lrenzyme0.6μl。25℃反应16h后,冰上静置5min。
产物采用热击法转化到大肠杆菌感受态trans5α中(具体操作方法同上)。pcr阳性鉴定方法同上,鉴定引物序列为:
正向引物:5’-gacacgctcgagatcacaag-3’
反正引物:5’-agaaagctgggttgtcgactgagaccggttgc-3’
获得阳性克隆后,将阳性克隆进行测序(由武汉擎科生物科技公司完成)。测序正确的克隆摇菌并使用北京全式金生物技术有限公司质粒小提试剂盒提取载体pearleygate101-ppcas1质粒。随后将该质粒转入农杆菌gv3101感受态中(唯地生物,中国)
农杆菌转化方法如下:
(1)取-80℃保存的农杆菌感受态于室温或手心片刻待其部分融化,处于冰水混合状态时插入冰中;
(2)每100μl感受态加入0.2-1μl质粒dna,用手拨打管底混匀,依次于冰上静置5min,液氮5min,37℃水浴5min,冰浴5min;
(3)加入700μl无抗生素的lb液体培养基,28℃摇床,220rpm培养2-3h;
(4)6000rpm离心1min收菌,留取100μl左右上清轻轻吹打重悬菌体涂布于含有相应抗生素的lb固体培养基上,培养皿倒置放于28℃恒温培养箱中,培养2-3d。
3.2采用烟草叶片瞬时转化的方法进行亚细胞定位
(1)将含有质粒pearleygate101-ppcas1和线粒体marker(mito-rfp)的农杆菌在添加相应抗生素的lb固体培养基上划线活化48h,挑取单克隆接种至3ml含有相应抗生素的lb液体培养基中,28℃摇床,220rpm培养过夜;
(2)取将过夜培养的菌液,以1%转接到含有相应抗生素的lb液体培养基中28℃摇床,220rpm培养过夜(约16h);
(3)将培养好的菌液4000rpm离心5min,去除上清,用悬浮液(10mmmgcl2,150μlacetosyringone,10mmmes,ph5.6)重悬菌体,测定od600值约1.0左右。pearleygate101-ppcas1(yfp)和mito-rfp(线粒体marker载体)等体积混合至终od600=0.5左右,室温避光静置4h;
(4)挑选移栽4周左右的本氏烟草,用1ml注射器将菌液注射到最上部3片全展功能叶整个叶片(从背面注射);
(5)注射2-3天后,取部分注射叶片在激光共聚焦显微镜(leica,德国)下观察荧光效果。
图4为ppcas1蛋白的亚细胞定位结果图。其中:图4a,
pearleygate101-ppcas1(yfp)在荧光灯下成像;图4b,线粒体marker(mito-rfp)在荧光灯下成像;图4c,明场;图4d,为pearleygate101-ppcas1(yfp)和线粒体marker(mito-rfp)叠加后的成像。通过图4可得该基因定位于线粒体。
四、烟草的遗传转化
1.植物转化载体构建
利用gateway方法经过两轮pcr反应构建pk7wg2d-ppcas1超表达载体。具体操作方法同亚细胞定位载体构建。
第一轮、第二轮pcr反应及第一次阳性鉴定引物序列同上;
通过bp反应将ppcas1序列连接至中间载体pdonr221上,测序正确后得到pdonr221-ppcas1质粒,再经过lr反应将目的基因序列连接至含有35s启动子的超表达载体pk7wg2d上,通过pcr反应鉴定阳性克隆,鉴定引物序列为:
正向引物:5’-cgcactagtgatatcacaag-3’
反正引物:5’-agaaagctgggttttagtcgactgagaccggttgc-3’
经过测序,获得pk7wg2d-ppcas1重组载体。
2.农杆菌介导的烟草遗传转化步骤如下(图5a-d)
(1)本氏烟草无菌苗准备:用2.0ml离心管取适量本氏烟种子,加1.5ml常温蒸馏水浸泡12-24h后洗去漂浮于水面不饱满的种子和杂质。在超净工作台中消毒以上处理过的种子。首先加入1.5ml75%医用酒精上下颠倒离心管45s,静置离心管,种子沉淀后用移液枪弃去酒精。加1.5ml无菌水上下颠倒2次,静置离心管,种子沉淀后用移液枪弃去水,此步重复两次;再加入1.5ml84消毒液上下颠倒离心管4min,静置离心管,种子沉淀后用移液枪弃去84消毒液。加1.5ml无菌水上下颠倒2次,静置离心管,种子沉淀后用移液枪弃去水,此步重复5次。最后把消毒好的种子均匀铺在ms平板上(培养基:1xmssalts;0.8%琼脂;3%蔗糖;ph5.7),吹干ms平板表面的水后封口放入25℃光照培养室培养。种子萌发后待其开始长出真叶时(大约2周)可将本氏烟草单株移到组培盒中培养。单株的烟草在组培盒中培养至大约4周时可用作侵染。
(2)菌液准备:用保存的菌液在含有相应抗生素lb固体平板上划线,平板在28℃恒温培养箱中培养36-48h,挑取单克隆摇菌并阳检。选取阳性克隆菌液转接至50ml含有相应抗生素的lb液体培养基中28℃200rpm摇菌12-16h直至菌液od值为0.5-0.7。
(3)侵染:从叶柄中间部位切下叶子,叶子背面朝下平铺在ms平板上(培养基:1xmssalts;0.8%琼脂;3%蔗糖;3μg/mlnaa;1μg/ml6-ba;ph5.7)尽量使叶子完全和培养基接触,但在此过程中尽量不要戳伤叶片。封口后平板正放在25℃黑暗条件下培养24-36h。在平皿中倒入适量的菌液(步骤2准备的菌液)。取出暗培养处理过的烟草叶片,在无菌滤纸上沿叶脉切成宽度为0.3cm的条状(切完的叶片呈羽毛状)。将切好的叶片浸没在菌液中,每5min摇晃一次培养皿,浸泡15-20min后取出叶片用无菌滤纸吸干叶片上的菌液。侵染完成的叶片继续放在ms平板上(培养基:1xmssalts;0.8%琼脂;3%蔗糖;3μg/mlnaa;1μg/ml6-ba;ph5.7),尽量使叶子与培养基充分接触,条状叶片不重叠,但在此过程中尽量不要戳伤叶片。封口后平板正放在25℃黑暗条件下培养48h。
(4)筛选培养:暗培养后,拿出叶片,从主叶脉上切下条状叶片放入筛选培养基上25℃光照培养(培养基:1xmssalts;0.8%琼脂;3%蔗糖;1μg/ml6-ba;50μg/ml硫酸卡那霉素;150μg/ml羧苄青霉素;ph5.7)。培养4天后,叶片开始转色,2周后开始出现愈伤组织,3周开始出现再生芽,这时可以将叶子转入新的培养基中继续培养(培养基:1xmssalts;0.8%琼脂;3%蔗糖;1μg/ml6-ba;50μg/ml硫酸卡那霉素;150μg/ml羧苄青霉素;ph5.7),促进生长。一月后可以切下再生芽生根。
(5)生根:把切下的芽竖直插入生根培养基中(培养基:1xmssalts;0.8%琼脂;3%蔗糖;150μg/ml羧苄青霉素;ph5.7)。2周后逐渐长出根,根长超出1cm后可以将再生苗移植到栽培基质中。
3.转基因阳性苗的筛选
(1)转基因烟草dna提取:取1/2指甲盖大小的叶片到2.0ml离心管中,装入1个钢珠,加入800μltps溶液(1ltps:1mtris-hcl;0.5medta;kcl74.5g;1%pvp40),研磨机70hz研磨2min;75℃水浴锅中加热1h,每20min翻转1次;水浴完成后将离心管12000rpm,离心10min,取上清500μl转移至新的1.5ml离心管中,加入等体积预冷的异丙醇,轻轻上下倒置几次混匀,-20℃静置1h;12000rpm,离心10min,弃上清,加入600μl75%乙醇离壁清洗;吸干残留的乙醇,风干后加入50μlddh2o溶解dna,-20℃保存备用。
(2)转基因烟草pcr鉴定
以提取的转基因烟草dna为模板,进行pcr鉴定。反应体系为:2×taqmastermix(dye)12.5μl,正向引物1μl,反向引物1μl,模板1μl,ddh2o9.5μl。
阳性鉴定引物序列如下:
正向引物:5’-tttcatttggagaggactcc-3’
反正引物:5’-taacgtgactcccttaattc-3’
pcr程序为:
以未做侵染转化的烟草叶片dna作为阴性对照,以ppcas1质粒为阳性对照。未进行侵染转化的烟草叶片dna不能扩增出目的条带,ppcas1质粒能扩增出目的条带。如图5e所示能扩增出目的条带的再生烟草植株被初步鉴定为阳性转基因烟草株系。
(2)转基因阳性烟草中ppcas1表达水平鉴定
按前文方法提取转基因阳性烟草植株叶片rna,反转录成cdna,以此cdna为模板进行半定量检测ppcas1表达水平。所用引物序列如下:
ppcas1-qpcr:
正向引物:5’-cttcctggaacccacgtcaa-3’
反向引物:5’-tccaccagtactgtcttgcc-3’
内参引物nbactin:
正向引物:5’-ggctctaccatgttcccagg-3’
反向引物:5’-ggtgctgagagaagccaagat-3’
pcr反应体系为:2×taqmastermix(dye)12.5μl,正向引物1μl,反向引物1μl,模板1μl,ddh2o9.5μl。
pcr反应程序:
以未做侵染转化的烟草叶片cdna作为阴性对照,1%琼脂糖电泳结果显示,阴性对照没有条带,转基因阳性烟草植株均有不同程度的条带呈现(图5f)。选取其中#5和#14两个转基因系进行后续试验。
五、ppcas1转基因烟草和桃ppcas1瞬时沉默株系的抗盐性分析
1.ppcas1转基因烟草抗盐性分析
为了鉴定ppcas1转基因烟草是否和抗盐胁迫有关,对转ppcas1基因株系及野生型(wt)进行氯化钠处理,观察处理前后的表型,同时测定相关生理指标。其中:图6a是野生型烟草种子和转基因烟草种子在1/2ms培养基和含有100mm氯化钠的1/2ms培养基上的发芽情况观察及统计。从图中可以看出在100mm氯化钠处理下,转基因烟草发芽率比野生型更高。图6b是30天大的烟草叶片在200mm氯化钠溶液中处理3天前后的表型。从图中可以看出野生型烟草叶片经过200mm氯化钠溶液处理后呈现出叶片漂白,褪绿的症状。而转基因烟草叶片症状较轻。图6c是30天大的烟草植株在300mm氯化钠处理前后表型,从图中可以看出经过300mm氯化钠处理后,转基因烟草较野生型烟草受到的伤害更小。图6d是30天大的烟草植株在300mm氯化钠处理后叶绿素含量的测定,由图中可见转基因烟草经过氯化钠处理后,叶绿素a、叶绿素b及总叶绿素含量更高。图6e是30天大的烟草植株在300mm氯化钠处理后电导率含量的测定,由图可见转基因烟草氯化钠处理后电导率更低,说明转基因烟草细胞膜受损伤的程度更小。图6f是30天大的烟草植株在300mm氯化钠处理后丙二醛含量的测定。由图可见转基因烟草经过300mm氯化钠处理后丙二醛含量更低,说明转基因烟草膜脂过氧化物含量与野生型相比更低,细胞受损更小。
2.利用病毒介导的vigs技术瞬时沉默桃中ppcas1的抗盐评价
图7是ppcas1沉默株系(ptrv-1,ptrv-2)及对照植株(wtp)盐处理前后表型观察及相关生理指标测定结果。其中:图7a是qrt-pcr检测ppcas1沉默株系(ptrv-1,ptrv-2)和对照植株(wtp)中ppcas1基因表达水平。图7b是20天大的ppcas1沉默株系(ptrv-1,ptrv-2)和对照植株(wtp)在300mm氯化钠处理前后表型,从图中可以看出经过300mm氯化钠处理后,ppcas1沉默株系较对照植株受到的伤害更大。图7c是20天大的ppcas1沉默株系(ptrv-1,ptrv-2)和对照植株(wtp)在300mm氯化钠处理后叶绿素含量的测定结果。图7d是20天大的ppcas1沉默株系(ptrv-1,ptrv-2)和对照植株(wtp)在300mm氯化钠处理后电导率测定结果,由图可见ppcas1沉默株系在盐处理后与对照植株相比,电导率更高,细胞膜受损伤更大。图7e是是20天大的ppcas1沉默株系(ptrv-1,ptrv-2)和对照植株(wtp)在300mm氯化钠处理后丙二醛含量测定结果,由图可见ppcas1沉默株系在300mm氯化钠处理后与对照植株相比产生的膜脂过氧化物更多,细胞受损更严重。
3.活性氧含量测定、组织化学染色分析h2o2和o2-积累及抗氧化酶活测定盐处理后,转基因烟草株系受到伤害的程度更小,电导率更低表明它们可能较wt可能积累更少的活性氧。相反,ppcas1沉默株系较对照株系可能积累更多的活性氧。那么通过鉴定植株中活性氧的积累量便成为必要。将材料进行300mm氯化钠溶液处理,取样测定组织中过氧化氢和抗超氧阴离子含量,并利用组织化学染色观察植株中活性氧积累情况。具体操作方法如下:
(1)利用bca法测定样品中蛋白浓度(微量酶标法)
使用南京建成公司的试剂盒,操作如下步骤所示操作:
混匀,37℃孵育30min后,准确吸取200μl各管反应液,用移液枪准确地加入到新的96孔酶标板中,使用酶标仪于波长562nm,测定各孔吸光度值。
总蛋白浓度(μg/ml)=(测定od值-空白od值)/(标准od值-空白od值)×标准品浓度(563μg/ml)×样本测试前稀释倍数
(2)h2o2含量的测定
使用南京建成公司的试剂盒,操作如下步骤所示操作:
盖上盖,涡旋仪震荡混匀,室温下3000rpm/min,离心5min,准确吸取200μl各管反应液,用移液枪准确地加入到新的96孔酶标板中,使用酶标仪于波长405nm,测定各孔吸光度值。
组织中h2o2含量(mmol/gprot)=(测定od值-空白od值)÷(标准od值-空白od值)×标准品浓度(163mmol/l)÷待测样本蛋白浓度(gprot/l)从图8a和图8b中可以看到,经过300mm氯化钠处理2周后相较于野生型(wt),转基因烟草株系(#5和#14)中过氧化氢含量更少,抗超氧阴离子含量更多。从图8e和图8f可以看出,经过300mm氯化钠处理10天后,ppcas1沉默株系(ptrv-1,ptrv-2)相较于对照(wtp)积累的过氧化氢和丙二醛更多,抗超氧阴离子更少。
(3)抗超氧阴离子自由基活性的测定
使用南京建成公司的试剂盒,操作如下步骤所示操作:
操作完成后用涡旋仪震荡混匀,于室温下静置反应10min,用移液枪吸取200μl各管反应液,将其加入到干净的96孔酶标板中,于波长550nm处,用酶标仪测定各孔吸光度值。
抗超氧阴离子活力单位(u/gprot)=(对照od值-测定od值)÷(对照od值-标准od值)×标准品浓度(0.15mg/ml)×1000ml÷待测样本蛋白浓度(gprot/l)
(4)二氨基联苯胺(dab)染色和氯化硝基四氮唑蓝(nbt)染色
二氨基联苯胺(dab)是最常用且敏感度最好的过氧化物酶显色底物,在过氧化物存在时,该试剂可与其反应生成棕色的不溶性沉淀,根据形成沉淀的数量及颜色深浅可用来判断h2o2的含量。氯化硝基四氮唑蓝(nbt)是碱性磷酸酶最佳的底物组合之一,在碱性磷酸酶的催化下,nbt会被o2-还原生成不溶性的深蓝色产物,而可以清除o2-的超氧化物歧化酶则可以降低nbt被还原的程度,表现出的症状为生成的蓝色产物较浅,因此nbt染色结果可用来检测o2-的含量。
图8c和图8d是30天大烟草植株经过两周后野生型植株和两个转基因株系的dab和nbt染色结果。图8g和图8h是20天大桃苗经过300mm氯化钠处理10天后对照植株和ppcas1沉默株系的dab和nbt染色结果。如图7c和图7d:盐胁迫后,用dab对植株叶片进行染色,转基因株系叶片出现褐色的叶面积明显比野生型更小,且颜色较淡;用nbt染色的叶片,转基因株系比野生型的蓝色更浅,面积更小。说明转基因株系在盐胁迫下较野生型积累更少的活性氧(h2o2和o2-)。如8g和图8h:300mm氯化钠处理10天后,dab染色结果显示ppcas1沉默株系较对照出现褐色的叶面积更大,且颜色较深;nbt染色结果显示ppcas1沉默株系比对照植株的蓝色更深,面积更大。说明经过盐胁迫处理,ppcas1沉默植株积累的活性氧更多。
(5)超氧化物歧化酶(sod)、过氧化物酶(pod)、过氧化氢酶(cat)的活力分析
使用南京建成公司的试剂盒,操作如下步骤所示操作:
超氧化物歧化酶(sod)
sod抑制率(%)=[(对照od值-对照空白od值)-(测定od值-测定空白od值)]÷[(对照od值-对照空白od值)]×100%
sod活力(u/mgprot)=sod抑制率÷50%×(反应体系÷稀释倍数)÷待测样本蛋白浓度(mgprot/ml)
过氧化物酶(pod)
混匀后,3500rpm离心10min,取上清液于波长420nm处,1cm光径双蒸水调零,测定各管吸光度值。
pod活力(u/mgprot)=[(测定od值-对照od值)÷(12×比色光径1cm)]×[(反应总体积ml)÷(取样量ml)]÷反应时间(30分钟)÷匀浆蛋白浓度(mgprot/ml)×1000
过氧化氢酶(cat)
混匀后,准确吸取200μl各管反应液,用移液枪准确地加入到新的96孔酶标板中,使用酶标仪于波长405nm,测定各孔吸光度值。
组织匀浆中cat活力(u/mgprot)=(对照od值-测定od值)×235.65×(1÷60÷取样量)÷待测样本蛋白浓度(mgprot/ml)
从图9a-c中可以看到,经过300mm氯化钠处理2周后相较于野生型(wt),转基因烟草株系(#5和#14)中、过氧化物酶(pod)、过氧化氢酶(cat)、超氧化物歧化酶(sod)活力更高。从图9d-f可以看出,经过300mm氯化钠处理10天后,ppcas1沉默株系(ptrv-1,ptrv-2)相较于对照(wtp)3种抗氧化酶活力更低。
综合分析表明,将ppcas1基因转入烟草后鉴定其功能,发现转基因超表达株系与对照野生型相比抗盐能力有了很大提升。转基因烟草中的过氧化氢(h2o2)、丙二醛(mda)以及超氧阴离子(o2-)的含量均比野生型要低,植株体内活性氧残留更少,细胞损伤更小,同时检测发现转基因烟草中超氧化物歧化酶(sod)、过氧化物酶(pod)、过氧化氢酶(cat)活力升高。而当桃中ppcas1基因被沉默后,这些指标表现出相反的结果。这些数据表明:过表达ppcas1基因能够有效的增强转基因植株的活性氧清除能力,从而提高了植株的耐盐性。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
序列表
<110>华中农业大学
<120>桃氰丙氨酸合成酶基因在提高植物耐盐性方面的应用
<160>1
<170>siposequencelisting1.0
<210>1
<211>1125
<212>dna
<213>桃(prunuspersica)
<400>1
atggcggccttgaggagctttctgaagaaaagatccactttgctctgcaatgaagccgtg60
agaaagagactcttctcaacccaagtgagccaacccatcattgattctccatcttttgct120
cagagggtcagaaaccttcctaaggatcttcctggaacccacgtcaaaacagaagtctcc180
caactcattggtagaactcccattgtctatctcaacaaagtcactgaaggatgtggagct240
tatatagctgtcaagcaagagatgtttcaacccacctctagcatcaaagacagaccagca300
ctttcaatgatcaacgatgcagaaaagaaaggcttgataactcctggcaagacagtactg360
gtggagccaacatcaggaaatatgggaatcagcatggcttttatggcagccatgagaggg420
tataaaatggttctcactatgccgtcttacacaagcttggagagaagggtgtgtatgaga480
gcctttggagctgaattaattctcactgatccagccagggggatgggaggaactgttaag540
aaggcttatgatcttctggaatccacaccaaatgctcatatgctccaacagttctcaaat600
cctgccaatactcgggtgcatttcgaaactacaggccctgagatatgggaggataccgat660
gggcaagttgacatcttcataatgggaattggtagcggaggcacagtctctggggttggg720
cagtatctcaaatccaaaaatcccaatgttcagatatatggagtggagcctgctgaaagt780
aatgtgctaaacggtggcaaaccaggtcctcatttgatcactggcaacggggttggattc840
aaaccagatattttggacttggatgtgatggatagagttattgaggttaaaagtgatgat900
gcagtaaaaatggcaagacgattggcattggaagagggacttatggtaggaatatcatcc960
ggagccaacacggtggcagcaattgagcttgctaaaaagccagaaaacaaaggcaaactt1020
attgtgaccgttcacccaagtttcggggagcgatacttgtcatctgtcctgtttgaagag1080
ctgaggaaggaagctgaaaacatgcaaccggtctcagtcgactaa1125
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



