
 商标分类
商标分类  商标转让
商标转让 
一种上/下转换双模式发光纳米晶及其制备方法和应用与流程
 2021-02-02 17:02:44|
2021-02-02 17:02:44| 420|
420| 起点商标网
起点商标网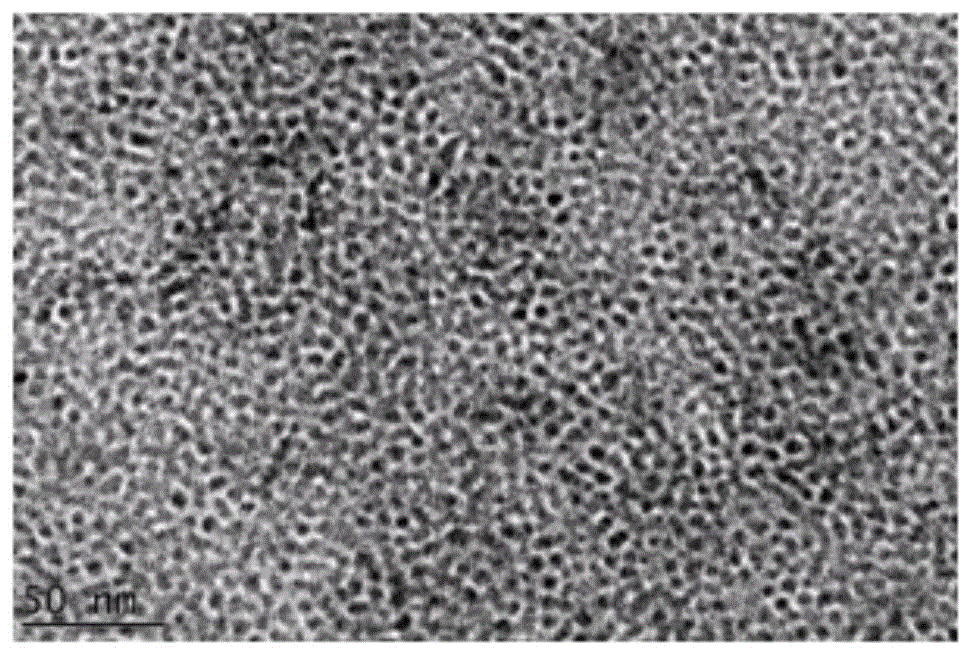
本发明涉及纳米材料技术领域,尤其涉及一种“葵花”状红光(发射峰约650nm)和近红外二区(发射峰约1520nm)上/下转换双模式发光纳米晶及其制备方法和应用。
背景技术:
上转换发光是一种反斯托克斯(anti-stokes)过程,指能够吸收两个或两个以上长波长光子而发射一个短波长光子的发光现象。稀土上转换发光材料的基质主要包括氧化物、氟化物、氟氧化物、含硫化合物、卤化物等,其中稀土氟化物具有声子能量低、无辐射跃迁少和元素可掺杂浓度高等特点,对上转换发光是较理想的基质,因此在发光材料领域具有广阔的应用前景。目前,研究最广泛的上转换发光材料为以er、tm或ho作为激活剂的稀土掺杂纳米晶。
近红外光激发的稀土上转换发光纳米技术因其独特的发光机理和优异的发光性能而在上转换光触发治疗等方面引起了广泛研究兴趣。稀土上转换纳米晶在应用于光触发治疗时具有较深的组织穿透深度、较低的自发荧光和优异的抗光漂白性能,因此比较适合用于深层生物组织中的“光子转换器”。然而,对于上转换荧光成像,由于上转换的短波长光子依然存在被生物组织高吸收和高散射等问题。此外,同时利用上转换荧光实现光触发治疗和光学成像存在能量分配不可控或能量供给不足的问题。因此,为了获得更高的光学成像信噪比,开发具有优异近红外(700-1700nm)荧光性能的稀土纳米晶极为必要。目前有课题组报道yb/ce/er或ce/er共掺杂纳米晶用于近红外二区生物传感或成像相关的研究,然而ce的高掺杂会不可避免地带来基质晶格缺陷,进而导致发光强度降低。
在抗癌领域,稀土上转换荧光介导的光触发治疗依然是当前的研究热点。对于稀土发光纳米晶,仅仅具有近红外下转换荧光性能存在局限性。为了实现其在生物诊疗及其他相关领域的拓展应用,同时赋予稀土发光纳米晶上转换和下转换近红外双模式荧光性能具有重要意义。而寻求新的具有上转换和下转换双模式发光性能的纳米晶作为潜在的抗癌诊疗纳米平台,仍然需要进一步研究探索。
综上,制备以近红外光作激发光源,具备较强上转换红色和下转换近红外二区双模式荧光的高性能稀土氟化物纳米晶的方案或路线还没有报道过。
技术实现要素:
本发明的目的在于提供一种上/下转换双模式发光纳米晶及其制备方法和应用,该纳米晶呈“葵花状”,且在红外区(发射峰约为650nm)和近红外区(发射峰约为1520nm)均可发光,从而解决现有技术中存在的前述问题。
为了实现上述目的,本发明采用的技术方案如下:
一种上/下转换双模式发光纳米晶,化学表达式为:nagdf4:yb,ce,ho,er
@nagdf4:yb,ce;其中@表示包裹。
一种上/下转换双模式发光纳米晶的制备方法,包括以下步骤:
s1,分别合成稀土金属元素钆、镱、铈、铒及钬的氯化物;
s2,分别合成含有稀土金属元素钆、镱、铈、铒及钬的油酸盐;
s3,采用高温溶剂热法制备nagdf4:54%yb/20%ce/1%ho/2%er核纳米晶:按物质的量分数计,分别称取23%的氯化钆、54%的氯化镱、20%的氯化铈、1%的氯化钬和2%的氯化铒置于反应容器中,再向所述反应容器内加入体积比为2:5的油酸和十八烯;在搅拌、抽真空状态下将反应溶液加热至100~110℃,待不再有气泡产生,关闭真空装置,通氮气保持10~20min,然后将反应溶液升温至160~180℃保持30~60min,随后自然冷却得到溶液a;
将10~15ml溶有稀土氯化物前驱体物质的量2~4倍的氢氧化钠和3~6倍氟化铵的甲醇溶液缓慢加入上述溶液a中并继续搅拌30~45min;在搅拌、抽真空状态下将反应体系加热至100~120℃,待不再有气泡产生,关闭真空装置,通氮气保持10~20min,然后升温至240~260℃保持40~50min,随后自然冷却降至室温;经乙醇和环己烷离心洗涤后即获得纳米颗粒,并将纳米颗粒保存在环己烷液体中;
s4,采用包覆法制备
nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce:将物质的量之和与步骤s3中相等且物质的量分数分别为50%油酸钆、20%油酸镱、30%油酸铈,与稀土油酸盐物质的量4~6倍的氟化钠和步骤s3中保存纳米颗粒的环己烷溶液混合,再加入体积比为1:1的油酸和十八烯;在搅拌、抽真空状态下加热至110~120℃,待不再有气泡产生,关闭真空装置,通氮气保持0.5~1h,然后升温至300~320℃反应1~1.5h,随后自然冷却降至室温;经乙醇和环己烷离心洗涤后即获得nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce。
优选地,步骤s1中所述分别合成稀土金属元素钆、镱、铈、铒及钬的氯化物具体为:
在室温下,取10~30mmol稀土氧化物和60~180mmol浓盐酸在容器中混合均匀,混合溶液在搅拌下缓慢加热到80~90℃,加入10~30ml蒸馏水并继续反应0.5~1h,将得到的溶液冷却并过滤得到澄清透明溶液,继续在80~90℃加热至溶液表面有氯化物晶体析出,转移至60~70℃烘箱烘干,得到的固体粉末为相应金属元素的氯化物;
优选地,步骤s2中所述的分别合成含有稀土金属元素钆、镱、铈、铒及钬的油酸盐具体步骤为:
取10~30mmol相应稀土氯化物、30~90mmol油酸钠、30~50ml蒸馏水、40~70ml乙醇和70~110ml正己烷加入到容器中,混合溶液在搅拌下加热到70℃,持续反应4h后,停止加热;冷却至室温后,将混合溶液倒入分液漏斗中进行分离,水洗后,取上层液体在80℃下水浴烘干,室温下放置直到得到固体蜡状物质,所述固体蜡状物质即为含有相应金属元素的油酸盐前驱体;
优选地,制备得到的nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce保存在环己烷液体中。
本发明的另一目的还在于将制得的上/下转换双模式发光纳米晶在生物成像探针或药物载体上的应用。
本发明的有益效果是:
本发明公开了一种上/下转换双模式发光纳米晶及其制备方法和应用,采用高温溶剂热法和外延生长法制备nagdf4:yb,ce,ho,er@nagdf4:yb,ce核壳结构纳米晶。这种制备方法有四个特点:
1.生成这种材料的制备方法包括两步高温溶剂热法,简单易行,所生成的纳米晶分散性好,粒径分布均匀。
2.内核掺入稀土离子钆和镱时,钆的掺杂是有利于得到六方晶相的核纳米晶,六方晶相有利于实现较强的荧光发射;
3.在核内掺杂低含量的钬,不仅对下转换近红外发光没有明显影响,同时铈与钬间的三个交叉驰豫过程赋予纳米晶上转换红光发射功能,壳层的包覆有利于提高纳米晶的上转换发光强度;
4.在尺寸较小的内核掺杂稀土离子铒和在尺寸较厚的壳内掺杂稀土离子铈时,由于铈和铒之间独特的能级匹配,当镱把能量传递给铒之后,铒和铈之间发生交叉驰豫效应,内核的小尺寸则非常有利于提高这种交叉驰豫的发生概率,进而导致铒的红光和绿光能级上光子急剧减少和近红外二区光能级上光子迅速累积;核内铒离子掺杂浓度为2%,镱离子掺杂浓度为传统的20%,其中壳层镱的掺杂大大提高了纳米晶对近红外激发光子的吸收能力;铒的近红外二区发光强度随着壳层铈离子掺杂浓度的增大先升后降,当铈的掺杂浓度为30%时近红外二区发射最强,比未掺杂ce的核壳结构纳米晶高了11.7倍。
本发明中的制备方法合成路线操作简单易行、粒径分布均匀、产物纯度高,且绿色环保,通过不同稀土元素掺杂、控制核壳结构中核与壳的尺寸赋予纳米晶较强的红色和近红外二区荧光,近红外荧光使得其可以作为潜在的光学成像造影剂,而红光发射使得制备的纳米晶可以作为纳米能量“转换器”而应用于光触发治疗。因此该纳米晶可以作为一种潜在的诊疗一体化纳米平台。
附图说明
图1是实施例1中步骤(3)高温溶剂热法制备的核纳米晶的透射电子显微镜图片;
图2是实施例1中步骤(4)延生长法连续包覆制备的核壳结构纳米晶的透射电子显微镜图片;
图3是实施例1中制备得到的核壳结构纳米晶的x射线衍射图谱及六方晶相nagdf4的标准卡片;
图4是980nm激光激发核壳结构纳米晶的可见上转换和近红外二区下转换发射光谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施方式仅仅用以解释本发明,并不用于限定本发明。
本发明中提供的可在上/下转换双模式发光纳米晶的化学表达式是nagdf4:yb,ce,ho,er@nagdf4:yb,ce;其中@表示包裹,在实际的制备生产中可以根据实际的需要对各个金属的含量进行调整。以下将给出几个实施例,用以制备出nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce。
实施例1
(1)分别合成氯化钆、氯化镱、氯化铈、氯化钬、氯化铒:在室温下,取20mmol相应金属氧化物和60mmol浓盐酸在容器中混合均匀,混合溶液在恒温加热磁力搅拌器上磁力搅拌下缓慢加热到80℃,加入10ml蒸馏水并继续反应0.5h,将得到的溶液冷却并过滤得到澄清透明溶液,继续在80℃加热直到溶液表面有氯化物晶体析出,转移至60℃烘箱烘干,得到的固体粉末为相应金属元素的氯化物;
(2)分别合成油酸钆、油酸镱和油酸铈:取20mmol相应金属氯化物、60mmol油酸钠、30ml蒸馏水、40ml乙醇和70ml正己烷加入容器中,混合溶液在恒温加热磁力搅拌器中磁力搅拌下加热到70℃,反应4h,停止加热冷却至室温后,将混合溶液倒入分液漏斗中,用蒸馏水洗三次,取上层液体在80℃下水浴烘干,在室温下放置三天,得到的固体蜡状物质为含有稀土元素的油酸盐前驱体;
(3)采用高温溶剂热法制备nagdf4:54%yb/20%ce/2%ho/0.5%er核纳米晶。称取0.0615g氯化钆、0.1519g氯化镱、0.0559g氯化铈、0.0055g氯化钬和0.0014g氯化铒于三口烧瓶中,加入6ml油酸和15ml十八烯;在搅拌、抽真空状态下加热至110℃,待不再有气泡产生,关闭真空装置,通氮气保持10min,然后升温至160℃保持30min,随后自然冷却降至室温;将10ml溶有0.1g氢氧化钠和0.1482g氟化铵的甲醇溶液缓慢加入上述溶液并继续搅拌30min;在搅拌、抽真空状态下将反应体系加热至120℃,待不再有气泡产生,关闭真空装置,通氮气保持10min,然后升温至250℃保持45min,随后自然冷却降至室温,经乙醇和环己烷洗涤三次后,所制备的纳米颗粒被保存在环己烷液体中;
(4)采用包覆法制备nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce。在100ml三口瓶中加入上一步的环己烷溶液;同时称取0.5008g油酸钆、0.2035g油酸镱、0.2954g油酸铈和0.21g氟化钠于三口烧瓶中,加入15ml油酸和15ml十八烯;在搅拌、抽真空状态下加热至120℃,待不再有气泡产生,关闭真空装置,通氮气保持20min,然后升温至300℃反应保持1h,随后自然冷却降至室温;经乙醇和环己烷洗涤三次后,所制备的纳米颗粒被保存在环己烷液体中。
实施例2
(1)分别合成氯化钆、氯化镱、氯化铈、氯化钬、氯化铒:在室温下,取30mmol相应金属氧化物和180mmol浓盐酸在容器中混合均匀,混合溶液在恒温加热磁力搅拌器上磁力搅拌下缓慢加热到90℃,加入30ml蒸馏水并继续反应1h,将得到的溶液冷却并过滤得到澄清透明溶液,继续在90℃加热直到溶液表面有氯化物晶体析出,转移至70℃烘箱烘干,得到的固体粉末为相应金属元素的氯化物;
(2)分别合成油酸钆、油酸镱和油酸铈:取30mmol相应金属氯化物、90mmol油酸钠、50ml蒸馏水、60ml乙醇和90ml正己烷加入容器中,混合溶液在恒温加热磁力搅拌器中磁力搅拌下加热到70℃,反应4h,停止加热冷却至室温后,将混合溶液倒入分液漏斗中,用蒸馏水洗三次,取上层液体在80℃下水浴烘干,在室温下放置三天,得到的固体蜡状物质为含有稀土元素的油酸盐前驱体;
(3)采用高温溶剂热法制备nagdf4:54%yb/20%ce/2%ho/0.5%er核纳米晶。称取0.0615g氯化钆、0.1519g氯化镱、0.0559g氯化铈、0.0055g氯化钬和0.0014g氯化铒于三口烧瓶中,加入6ml油酸和15ml十八烯;在搅拌、抽真空状态下加热至100℃,待不再有气泡产生,关闭真空装置,通氮气保持20min,然后升温至180℃保持45min,随后自然冷却降至室温;将15ml溶有0.1g氢氧化钠和0.1482g氟化铵的甲醇溶液缓慢加入上述溶液并继续搅拌45min;在搅拌、抽真空状态下将反应体系加热至110℃,待不再有气泡产生,关闭真空装置,通氮气保持20min,然后升温至240℃保持50min,随后自然冷却降至室温,经乙醇和环己烷洗涤三次后,所制备的纳米颗粒被保存在环己烷液体中;
(4)采用包覆法制备nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce。在100ml三口瓶中加入上一步的环己烷溶液;同时称取0.5008g油酸钆、0.2035g油酸镱、0.2954g油酸铈和0.21g氟化钠于三口烧瓶中,加入15ml油酸和15ml十八烯;在搅拌、抽真空状态下加热至110℃,待不再有气泡产生,关闭真空装置,通氮气保持30min,然后升温至310℃反应保持1.5h,随后自然冷却降至室温;经乙醇和环己烷洗涤三次后,所制备的纳米颗粒被保存在环己烷液体中。
实施例3
(1)分别合成氯化钆、氯化镱、氯化铈、氯化钬、氯化铒:在室温下,取10mmol相应金属氧化物和90mmol浓盐酸在容器中混合均匀,混合溶液在恒温加热磁力搅拌器上磁力搅拌下缓慢加热到85℃,加入20ml蒸馏水并继续反应0.8h,将得到的溶液冷却并过滤得到澄清透明溶液,继续在85℃加热直到溶液表面有氯化物晶体析出,转移至65℃烘箱烘干,得到的固体粉末为相应金属元素的氯化物;
(2)分别合成油酸钆、油酸镱和油酸铈:取10mmol相应金属氯化物、30mmol油酸钠、40ml蒸馏水、70ml乙醇和110ml正己烷加入容器中,混合溶液在恒温加热磁力搅拌器中磁力搅拌下加热到70℃,反应4h,停止加热冷却至室温后,将混合溶液倒入分液漏斗中,用蒸馏水洗三次,取上层液体在80℃下水浴烘干,在室温下放置三天,得到的固体蜡状物质为含有稀土元素的油酸盐前驱体;
(3)采用高温溶剂热法制备nagdf4:54%yb/20%ce/2%ho/0.5%er核纳米晶。称取0.0615g氯化钆、0.1519g氯化镱、0.0559g氯化铈、0.0055g氯化钬和0.0014g氯化铒于三口烧瓶中,加入6ml油酸和15ml十八烯;在搅拌、抽真空状态下加热至105℃,待不再有气泡产生,关闭真空装置,通氮气保持15min,然后升温至170℃保持60min,随后自然冷却降至室温;将12ml溶有0.1g氢氧化钠和0.1482g氟化铵的甲醇溶液缓慢加入上述溶液并继续搅拌40min;在搅拌、抽真空状态下将反应体系加热至100℃,待不再有气泡产生,关闭真空装置,通氮气保持15min,然后升温至260℃保持40min,随后自然冷却降至室温,经乙醇和环己烷洗涤三次后,所制备的纳米颗粒被保存在环己烷液体中;
(4)采用包覆法制备nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce。在100ml三口瓶中加入上一步的环己烷溶液;同时称取0.5008g油酸钆、0.2035g油酸镱、0.2954g油酸铈和0.21g氟化钠于三口烧瓶中,加入15ml油酸和15ml十八烯;在搅拌、抽真空状态下加热至110℃,待不再有气泡产生,关闭真空装置,通氮气保持60min,然后升温至320℃反应保持1.2h,随后自然冷却降至室温;经乙醇和环己烷洗涤三次后,所制备的纳米晶颗粒被保存在环己烷液体中。
选用实施例1中制得的产物进行形态表征,对于第三步中制得的核纳米晶投射电子显微镜图片如图1所示,对于第四步得到的核壳结构纳米晶其表征结果如
上述三个实施例中记载的制备方法有四个特点:一是生成这种材料的制备方法包括两步高温溶剂热法,简单易行,所生成的纳米晶分散性好,粒径分布均匀。二是内核掺入稀土离子钆和镱时,钆的掺杂是有利于得到六方晶相的核纳米晶,六方晶相有利于实现较强的荧光发射;三是在核内掺杂低含量的钬,不仅对下转换近红外发光没有明显影响,同时铈与钬间的三个交叉驰豫过程赋予纳米晶上转换红光发射功能,壳层的包覆有利于提高纳米晶的上转换发光强度。四是在尺寸较小的内核掺杂稀土离子铒和在尺寸较厚的壳内掺杂稀土离子铈时,由于铈和铒之间独特的能级匹配,当镱把能量传递给铒之后,铒和铈之间发生交叉驰豫效应,内核的小尺寸则非常有利于提高这种交叉驰豫的发生概率,进而导致铒的红光和绿光能级上光子急剧减少和近红外二区光能级上光子迅速累积;核内铒离子掺杂浓度为2%,镱离子掺杂浓度为传统的20%,其中壳层镱的掺杂大大提高了纳米晶对近红外激发光子的吸收能力;铒的近红外二区发光强度随着壳层铈离子掺杂浓度的增大先升后降,当铈的掺杂浓度为30%时近红外二区发射最强,比未掺杂ce的核壳结构纳米晶高了11.7倍。
采用上述实施例中得到的纳米晶,可用于制备药物载体,比如介孔纳米药物载体,制备方法如下:
将实施例1-3中任意制得的纳米晶与十六烷基三甲基溴化铵和去离子水混合,搅拌至透明、澄清溶液;加入乙醇和氢氧化钠溶液;混合物在搅拌状态下加热到70~75℃,逐滴加入正硅酸乙酯,反应过程持续10~15min;用乙醇反复洗涤后,加入乙醇和硝酸铵,加热至60℃并保持2~3h后,用乙醇离心分离,所得产物即为包覆介孔二氧化硅后的药物载体,具体化学表达式为:
nagdf4:54%yb/20%ce/2%ho/0.5%er@nagdf4:20%yb/30%ce@msio2。
通过采用本发明公开的上述技术方案,得到了如下有益的效果:
本发明中提供的合成路线操作简单易行、粒径分布均匀、产物纯度高,且绿色环保,通过不同稀土元素掺杂、控制核壳结构中核与壳的尺寸赋予纳米晶较强的红色和近红外二区荧光,近红外荧光使得其可以作为潜在的光学成像造影剂,而红光发射使得制备的纳米晶可以作为纳米能量“转换器”而应用于光触发治疗。因此该种纳米晶可用在生物成像探针或药物载体等领域,可以作为一种潜在的诊疗一体化纳米平台。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



