
 商标分类
商标分类  商标转让
商标转让 
6-(环丙烷甲酰氨基)-4-((2-甲氧基-3-(1-甲基-1H-1,2,4-三唑-3-基)苯基)氨基)-N-(甲基-D3)哒嗪-3-甲酰胺的结晶形式的制作方法
 2021-02-02 17:02:33|
2021-02-02 17:02:33| 338|
338| 起点商标网
起点商标网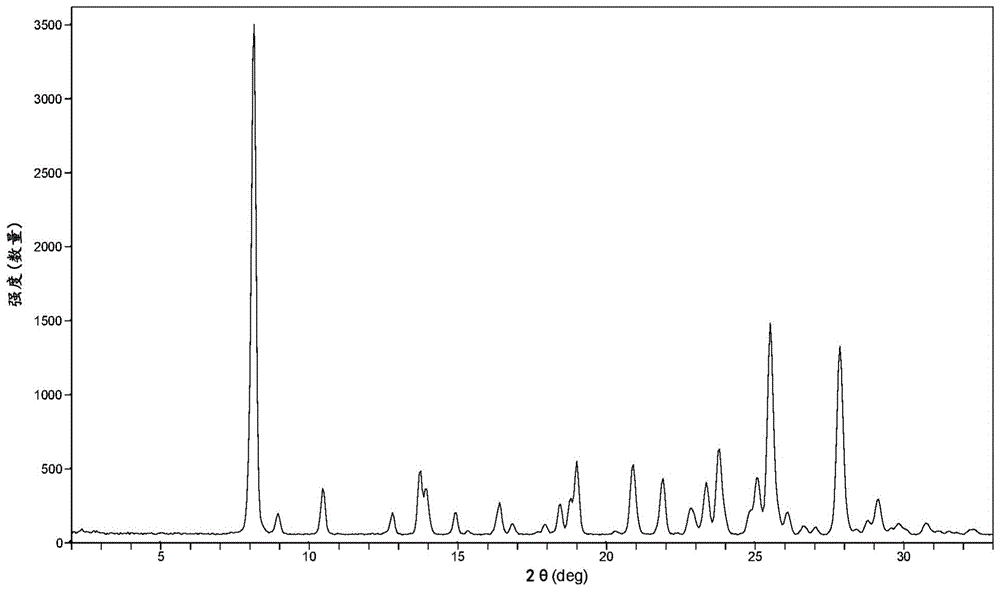
相关申请的交叉引用本申请要求2018年5月31日提交的美国临时申请号62/678451的权益,将其公开内容通过引用以其整体并入本文。本发明通常涉及6-(环丙烷甲酰氨基)-4-((2-甲氧基-3-(1-甲基-1h-1,2,4-三唑-3-基)苯基)氨基)-n-(甲基-d3)哒嗪-3-甲酰胺盐酸盐的结晶形式,下文称为“形式b”。
背景技术:
:化合物6-(环丙烷甲酰氨基)-4-((2-甲氧基-3-(1-甲基-1h-1,2,4-三唑-3-基)苯基)氨基)-n-(甲基-d3)哒嗪-3-甲酰胺具有式(i)的结构:并且在本文中称为“化合物(i)”。化合物(i)披露于美国专利9,505,748b2中,所述专利已转让给本受让人。美国专利9,505,748b2也披露了使用化合物(i)的治疗方法。化合物(i)是一种目前正在临床试验中用于治疗自身免疫疾病和自身炎性疾病的tyk2抑制剂,所述自身免疫疾病和自身炎性疾病例如银屑病、银屑病性关节炎、狼疮、狼疮性肾炎、舍格伦综合征(syndrome)、炎性肠病、克罗恩病(crohn’sdisease)和强直性脊柱炎。在合成旨在用于药物用途的化合物时,有必要在合成过程完成时且在进一步加工以提供呈药物配制品形式的化合物之前分离和纯化所述化合物。可以以组合或分开的连续步骤进行的分离和纯化步骤在从反应混合物的其他组分分离的过程中和/或在从分离的化合物样品去除杂质的纯化过程中以最小的产率损失提供化合物。希望提供可以从分离和/或纯化步骤可重复生产的固体形式。此外,希望分离呈固体形式的纯化的化合物,所述固体形式在一系列的储存条件下(如在不同的温度和湿度条件下)在物理上和化学上是稳定的。此外,还希望提供呈固体形式的化合物,所述固体形式适合于另外的加工,例如其中可以将其转化成其他固体形式(如无定型形式)。此外,希望提供呈固体形式的化合物,所述固体形式在溶剂/溶液中具有足够的溶解度以允许制备其他固体形式。申请人已经发现化合物(i)的结晶形式,其令人惊讶地提供了呈固体形式的化合物(i),所述固体形式在一系列储存条件下在物理上和化学上是稳定的。此外,申请人已经发现化合物(i)的结晶形式,其令人惊讶地提供了呈固体形式的化合物(i),所述固体形式在一系列储存条件下在物理上和化学上是稳定的,并且在溶剂/溶液中具有足够的溶解度以允许制备其他固体形式。此外,申请人已经发现化合物(i)的结晶形式,其令人惊讶地提供了呈固体形式的化合物(i),所述固体形式比其他测试的盐更好地减轻ph效应。本发明还涉及其他重要方面。技术实现要素:本发明提供了化合物(i)的结晶形式b。本文中用来表征特定形式的名称(例如“形式b”等)不应视为对具有相似或相同的物理和化学特征的任何其他物质进行限制,而是应该理解为该名称仅仅是标识符,其应根据本文另外提出的特征信息来解释。附图说明图1显示了观察到的化合物(i)的结晶形式b的粉末x射线衍射图(cukα,在t=25℃)。图2显示了化合物(i)的结晶形式b的差示扫描量热法(dsc)热谱图。图3显示了化合物(i)的形式b的热重分析(tga)热谱图。图4显示了在25℃下测量的化合物(i)的形式b的吸湿等温线。图5显示了化合物(i)的形式b的13c固态核磁共振(ssnmr)谱。具体实施方式在阅读下面详细描述时,本领域普通技术人员可以更容易地理解本发明的特征和优点。应当理解,出于清楚的原因,在单独的实施方案的上下文中描述的本发明的某些特征也可以组合以形成单个实施方案。相反,出于简洁的原因,在单个实施方案的上下文中描述的本发明的各种特征也可以被组合以形成其子组合。本文用来表征特定形式的名称,例如“形式b”等,仅是根据本文提供的表征信息进行解释的标识符,并且不应受限制以排除具有相似或相同物理和化学特征的任何其他物质。本文阐述的定义优先于通过引用并入本文的任何专利、专利申请和/或专利申请出版物中阐述的定义。所有在单词“约”前面表示成分的数量、重量百分比(wt%)、温度等的数字均应仅理解为近似值,因此可以使用高于和低于所述规定数字的微小变化来实现与所述规定数字基本相同的结果。因此,除非相反地指示,否则在单词“约”之前的数字参数是近似值,其可以根据试图获得的所需的特性而变化。至少并且不试图限制将等同原则应用于权利要求的范围,每个数字参数至少应被解释为是根据所报告的有效数字的数量并且通过应用普通修约技术的。所有测量都受到实验误差的影响,并且在本发明的精神之内。如本文所使用的,“多晶型物”是指具有相同化学结构但形成晶体的分子和/或离子的空间排列不同的晶体形式。如本文所使用的,“无定形”是指非结晶的分子和/或离子的固体形式。无定形固体不会展示出具有尖尖的极大值的确定的x射线衍射图。如本文所使用的,“基本上纯的”当关于结晶形式使用时意指具有如下纯度的化合物:基于化合物的重量,大于90重量%,包括大于90重量%、91重量%、92重量%、93重量%、94重量%、95重量%、96重量%、97重量%、98重量%和99重量%,并且还包括等于化合物(i)的约100重量%。其余材料包括化合物的其他一种或多种形式和/或由其制备产生的反应杂质和/或加工杂质。例如,化合物(i)的结晶形式可以被认为是基本上纯的,因为它具有大于90重量%的纯度,如通过当时在本领域中已知和普遍接受的手段所测量的,其中其余的小于10重量%的材料包含化合物(i)的无定形和/或其他一种或多种形式和/或反应杂质和/或加工杂质。如本文所使用的,“包含”选自特定峰组的多个峰的粉末x射线衍射(pxrd)图旨在包括具有不包含在特定峰组中的另外的峰的pxrd图。例如,包含四个或更多个、优选五个或更多个选自a、b、c、d、e、f、g和h的2θ值的pxrd图旨在包括具有以下项的pxrd图:(a)四个或更多个、优选五个或更多个选自a、b、c、d、e、f、g和h的2θ值;(b)零个或多个峰,其不是a、b、c、d、e、f、g和h峰中的任一个。反应杂质和/或加工杂质的存在可以通过本领域已知的分析技术来确定,所述分析技术例如色谱法、核磁共振波谱法、质谱法和/或红外光谱法。如本文所使用的,晶胞参数“每个晶胞中的分子”是指晶胞中化合物(i)的分子数。实施例(i)的形式b在一个实施方案中,化合物(i)作为包含形式b的结晶材料提供。化合物(i)的结晶形式b是hcl盐。在一个实施方案中,化合物(i)的结晶形式b的特征在于晶胞参数近似等于以下:α=114.3±0.5°β=94.1±0.5°γ=93.31±0.5°空间群:p-1每个晶胞中的分子(z):2计算密度1.424g/cm3其中所述的化合物(i)的形式b的晶胞参数是在约25°℃的温度下测量的。表1.化合物(i)的形式b选择的以度为单位的pxrd2θ值(cukα)8.1±0.28.9±0.210.4±0.212.8±0.214.9±0.216.4±0.216.8±0.217.9±0.218.4±0.220.8±0.221.8±0.222.8±0.223.3±0.223.8±0.224.8±0.225.1±0.225.4±0.226.1±0.226.6±0.227.0±0.227.8±0.228.4±0.228.8±0.229.1±0.229.6±0.229.9±0.230.7±0.2表2.化合物(i)的形式b的13cssnmr化学位移值在一个实施方案中,化合物(i)的结晶形式b的特征在于粉末x射线衍射图,其包含选自以下的四个或更多个以度为单位的2θ值(cukα):8.1±0.2;8.9±0.2;10.4±0.2;12.8±0.2;14.9±0.2;16.4±0.2;和18.4±0.2,其中形式b的pxrd图是在约25℃的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于粉末x射线衍射图,其包含选自以下的五个或更多个以度为单位的2θ值(cukα):8.1±0.2;8.9±0.2;10.4±0.2;12.8±0.2;14.9±0.2;16.4±0.2;和18.4±0.2,其中形式b的pxrd图是在约25℃的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于粉末x射线衍射图,其包含选自以下的六个或更多个以度为单位的2θ值(cukα):8.1±0.2;8.9±0.2;10.4±0.2;12.8±0.2;14.9±0.2;16.4±0.2;和18.4±0.2,其中形式b的pxrd图是在约25℃的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于粉末x射线衍射图,其包含等于8.1±0.2和12.8±0.2的以度为单位的2θ值(cukα);以及选自以下的三个或更多个以度为单位的2θ值(cukα):8.9±0.2;10.4±0.2;14.9±0.2;16.4±0.2和18.4±0.2;其中形式b的pxrd图是在约25℃的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于13cssnmr谱,其包含选自以下的四个或更多个以ppm为单位的化学位移值(均±0.2):175.0;167.6;156.3;150.9;147.2;144.4;142.7;138.2;136.3;125.0;121.6;117.5;98.3;59.8;40.6;26.9;15.6;10.3和7.2;其中形式b的光谱是在约280°k的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于13cssnmr谱,其包含选自以下的五个或更多个以ppm为单位的化学位移值(均±0.2):175.0;167.6;156.3;150.9;147.2;144.4;142.7;138.2;136.3;125.0;121.6;117.5;98.3;59.8;40.6;26.9;15.6;10.3和7.2;其中形式b的光谱是在约280°k的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于13cssnmr谱,其包含选自以下的六个或更多个以ppm为单位的化学位移值(均±0.2):175.0;167.6;156.3;150.9;147.2;144.4;142.7;138.2;136.3;125.0;121.6;117.5;98.3;59.8;40.6;26.9;15.6;10.3和7.2;其中形式b的光谱是在约280°k的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于13cssnmr谱,其包含等于117.5和40.6的以ppm为单位的化学位移值(均±0.2);以及选自以下的三个或更多个以ppm为单位的化学位移值:175.0;167.6;156.3;150.9;147.2;144.4;142.7;138.2;136.3;125.0;121.6;98.3;59.8;26.9;15.6;10.3和7.2;其中形式b的光谱是在约280°k的温度下测量的。在一个实施方案中,化合物(i)的结晶形式b的特征在于:(i)粉末x射线衍射图,其包含等于8.1±0.2和12.8±0.2的以度为单位的2θ值(cukα),在约25℃的温度下测量;和(ii)在约203℃至约217℃的近似范围内的宽吸热(broadendotherm)。在一个实施方案中,化合物(i)的结晶形式b的特征在于基本上如图1所示的观察到的粉末x射线衍射图。在一个实施方案中,化合物(i)的结晶形式b的特征在于在约203℃至约217℃的近似范围内的宽吸热。在一个实施方案中,化合物(i)的结晶形式b的特征在于基本上如图2中所示的差示扫描量热法(dsc)热谱图。在一个实施方案中,化合物(i)的结晶形式b的特征在于(i)粉末x射线衍射图,其包含等于8.1±0.2和12.8±0.2的以度为单位的2θ值(cukα),在约25℃的温度下测量;和(ii)基本上如图2中所示的差示扫描量热法(dsc)热谱图。在一个实施方案中,化合物(i)的结晶形式b的特征在于热重分析(tga)热谱图,其在加热至约150℃的温度时具有基于形式b的样品的重量的小于0.2%的重量损失。在一个实施方案中,化合物(i)的结晶形式b展现出基本上如图3所示的热重分析(tga)热谱图。在一个实施方案中,化合物(i)的结晶形式b展现出基本上如图4所示的吸湿等温线。在一个实施方案中,化合物(i)的结晶形式b的特征在于基本上如图5所示的固态nmr(ssnmr)。在仍甚至另外的实施方案中,化合物(i)的结晶形式b是基本上纯的。在另一个实施方案中,化合物(i)的结晶形式基本上由形式b组成。该实施方案的结晶形式可以包含基于化合物(i)的结晶形式即形式b的重量的至少约90wt%、优选至少约95wt%、以及更优选至少约99wt%。一个实施方案提供了一种包含6-(环丙烷甲酰氨基)-4-((2-甲氧基-3-(1-甲基-1h-1,2,4-三唑-3-基)苯基)氨基)-n-(甲基-d3)哒嗪-3-甲酰胺的组合物,其中至少95wt%、优选至少97wt%、以及更优选至少99wt%的所述6-(环丙烷甲酰氨基)-4-((2-甲氧基-3-(1-甲基-1h-1,2,4-三唑-3-基)苯基)氨基)-n-(甲基-d3)哒嗪-3-甲酰胺呈结晶形式b。可以通过多种方法来制备结晶形式,所述多种方法包括例如从合适的溶剂结晶或重结晶、升华、从熔体生长、从另一相进行固态转化、从超临界流体结晶以及喷射喷雾。用于从溶剂混合物结晶或重结晶的技术包括例如蒸发溶剂、降低溶剂混合物的温度、向分子和/或盐的过饱和溶剂混合物中添加晶种、将溶剂混合物冷冻干燥以及在溶剂混合物中添加抗溶剂(反溶剂)。可以采用高通量结晶技术来制备包括多晶型物的结晶形式。药物的晶体(包括多晶型物)、药物晶体的制备方法和表征在solid-statechemistryofdrugs,s.r.byrn,r.r.pfeiffer,和j.g.stowell,第2版,ssci,西拉斐特,印第安纳州(1999)中进行了讨论。对于采用溶剂的结晶技术,一种或多种溶剂的选择通常取决于一种或多种因素,例如化合物的溶解度、结晶技术和溶剂的蒸气压。可以使用溶剂的组合,例如,可以将化合物溶解到第一溶剂中以提供溶液,接着添加抗溶剂以降低化合物在溶液中的溶解度并提供晶体的形成。抗溶剂是在其中化合物具有低溶解度的溶剂。在一种制备晶体的方法中,将化合物在合适的溶剂中悬浮和/或搅拌以提供浆料,可以将其加热以促进溶解。如本文所使用的,术语“浆料”意指化合物的饱和溶液,其还可以含有另外的量的化合物以在给定温度下提供化合物和溶剂的不均匀混合物。可以将晶种添加至任何结晶混合物中以促进结晶。可以使用晶种来控制特定多晶型物的生长或控制晶体产物的粒度分布。因此,所需种子的量的计算取决于可用种子的大小和所需的平均产物颗粒的大小,如例如在“programmedcoolingofbatchcrystallizers,”j.w.mullin和j.nyvlt,chemicalengineeringscience,1971,26,369-377中所述。通常,需要小尺寸的种子来有效控制批料中晶体的生长。小尺寸的种子可以通过大晶体的筛分、研磨或微粉化,或通过溶液的微晶化来产生。应当注意,晶体的研磨或微粉化不会导致与所需的晶体形式相比发生任何结晶度形式变化(即变为无定形的或另一种多晶型物)。冷却的结晶混合物可以在真空下过滤,并且分离的固体可以用合适的溶剂(如冷的重结晶溶剂)洗涤,并且在氮气吹扫下干燥以提供所需的结晶形式。分离的固体可以通过合适的光谱或分析技术(如固态核磁共振、差示扫描量热法、粉末x射线衍射等)进行分析,以确保形成产物的优选结晶形式。基于结晶过程中最初使用的化合物的重量,通常以大于约70重量%的分离产率、优选大于90重量%的分离产率的量产生所得晶形。如果需要,可以将产物共研磨或通过网筛以切割产物。结晶形式可以直接从制备化合物(i)的最后过程的反应介质制备。例如,这可以通过在最后的过程步骤中采用可以使化合物(i)从中结晶的溶剂或溶剂混合物来实现。可替代地,可以通过蒸馏或溶剂添加技术获得结晶形式。用于此目的的合适溶剂包括例如上述非极性溶剂和极性溶剂,包括质子极性溶剂(如醇)和非质子极性溶剂(如酮)。样品中多于一种多晶型物的存在可以通过例如粉末x射线衍射(pxrd)或固态核磁共振(ssnmr)光谱法等技术确定。例如,在将实验测量的pxrd图与模拟的pxrd图进行比较时,额外的峰的存在可能指示样品中的多于一种多晶型物。可以从单晶x射线数据计算模拟的pxrd,请参见mith,d.k.,“afortranprogramforcalculatingx-raypowderdiffractionpatterns,”lawrenceradiationlaboratory,利弗莫尔,利福尼亚州,ucrl-7196(1963年4月)。化合物(i)的形式b可以使用多种技术来表征,其操作是本领域普通技术人员熟知的。可以使用单晶x射线衍射来表征和区分这些形式,这是基于在固定分析温度下单晶的晶胞测量。在stout和jensen,x-raystructuredetermination:apracticalguide,macmillanco.,纽约(1968),第3章中提供了晶胞的详细描述,将其通过引用并入本文。可替代地,表征晶体结构的另一种手段是通过粉末x射线衍射分析(其中将衍射曲线与代表纯粉末材料的模拟曲线进行比较,两者均在相同的分析温度下运行)和对表征为一系列以度为单位的2θ值(通常为四个或更多个)的主题形式的测量。可以使用表征所述形式的其他手段,如固态核磁共振、差示扫描量热法、热分析和振动光谱法。这些参数也可以组合使用来表征主题形式。效用化合物(i)的结晶形式b可以用于在合成过程完成时将化合物(i)与其他组分分离;和/或用于通过一个或一系列结晶步骤纯化化合物(i)。分离和纯化步骤可以组合或作为单独的过程步骤实施。实施例现在将通过以下一个或多个工作实施例进一步描述本发明,所述工作实施例是本发明的优选实施方案。除非另有说明,否则所有温度均以摄氏度(℃)为单位。这些实施例是说明性的而不是限制性的,并且应当理解,可能存在落入由所附权利要求所限定的本发明的精神和范围内的其他实施方案。为了便于引用,在本文中可以使用以下缩写。缩写实施例1:化合物(i)的结晶形式b的制备通过将51mg化合物(i)和0.5ml0.25mhcl混合到2mlipa中并加热到55℃来制备溶液。将混合物在55℃下搅拌过夜,然后关闭加热,并在室温下搅拌过夜,得到化合物(i)的形式b的浓浆料。实施例2:化合物(i)的结晶形式b的制备通过在室温(25℃)下将550mg化合物(i)混合到35mlthf和2ml水中直至完全溶解,在其中添加108μl37%hcl来制备溶液。使用speedvac将所得浆料干燥过夜。将干燥的固体在60℃下悬浮于5ml的buoac中,并将所得的浆料在60℃下老化过夜。将浆料过滤并将湿滤饼在真空烘箱中在50℃-60℃的温度下干燥,以提供化合物(i)的形式b。实施例3:化合物(i)的结晶形式b的制备通过在60℃下混合395μl37%hcl(1.0摩尔当量)、25mlbuoac和7.6mldma制备hcl溶液。在60℃下分别将2g化合物i和15mldma装入100ml三颈圆底烧瓶中,得到澄清溶液,向其中添加3mlhcl溶液以保持澄清溶液。添加40mg化合物(i)形式b的种子后产生浑浊溶液,然后在3小时内用注射泵添加剩余的hcl溶液。在1小时内将得到的浆料从60℃冷却至50℃,并在50℃下将其搅拌15小时。然后在1小时内将浆料冷却至20℃,并搅拌过夜。然后将冷却的浆料过滤,并将湿滤饼用6ml的buoac洗涤。将湿滤饼在真空烘箱中在50℃下干燥过夜,提供2.1g化合物(i)hcl盐形式b(93%产率)。实施例4:化合物(i)的结晶形式b的制备通过装入1.4kg的化合物(i)crude和8.400l的nmp,并加热至50℃制备溶液。将得到的溶液在50℃下在celite545床上精制过滤。用2.5l的nmp冲洗溶解容器两次,并且每次冲洗也都经过精制过滤。将溶液加热至50℃并添加0.421kg盐酸37wt%溶液,以在溶液中形成化合物(i)hcl盐。在30分钟内将14l乙酸乙酯装入批料中以形成形式b浆料。在60分钟内将浆料冷却至20℃。将冷却的浆料老化12小时,然后将其过滤。用2×5.6l乙酸乙酯洗涤湿滤饼。将湿滤饼放入真空烘箱中并在55℃下干燥,以提供1.3kg呈形式b的化合物(i)(98.95ap,85.2%产率)。实施例5:化合物(i)的结晶形式b的制备通过装入2.323kg的化合物(i)crude和16.26l的nmp并加热至50℃来制备溶液。将所得溶液在50℃下精制过滤。用2.32lnmp冲洗溶解容器,并且将冲洗也进行精制过滤。将该溶液加热至50℃并添加0.55kg盐酸37wt%溶液,以在溶液中形成化合物(i)hcl盐。将0.04kg的化合物(i)的形式b和0.47l的乙酸乙酯装入批料中以产生浆料。将得到的浆料老化90分钟,然后在60分钟内将13.48l乙酸乙酯装入批料中。将浆料在50℃下老化60分钟,然后在60分钟内将浆料冷却至20℃。将冷却的浆料老化12小时,然后将其过滤。用2×9.29l乙酸乙酯洗涤湿滤饼。将湿滤饼放入真空烘箱中,并在65℃下干燥,以提供2.27kg呈形式b的化合物(i)(99.93ap,89.1%产率)。6-(环丙烷甲酰氨基)-4-((2-甲氧基-3-(1-甲基-1h-1,2,4-三唑-3-基)苯基)氨基)-n-(甲基-d3)哒嗪-3-甲酰胺的合成步骤1:化合物2的制备向搪玻璃反应器中装入甲苯(0.26kg)、环丁砜(3.4kg)、化合物1(1.0kg)和pocl3(2.7kg)。将粗品冷却至0℃。装入三乙胺(0.89kg),并将所得的粗混合物加热至65℃并老化直至反应完成。将反应物料冷却至5℃。在单独的反应器中,装入水(7.5kg)并冷却至5℃。将反应物料缓慢添加至水溶液中,保持内部温度低于5℃。使用另外的水(0.5kg)冲洗反应器,并帮助转移。将得到的混合物在5℃下搅拌3小时,然后用mtbe萃取3次(3×4.5kg)。依次用ph7缓冲液(5.0l/kg,15wt%kh2po4/k2hpo4)和水(2.5kg)洗涤合并的有机层。在真空下蒸馏粗品,直到总体积变为约3l/kg。添加acn(2×6.3kg),接着再蒸馏回约3l/kg。将粗品冷却至20℃以提供作为30wt%-36wt%溶液的化合物2,产率为90%-95%。步骤2:化合物3的制备在25℃下将acn(2.7kg)、溴化锂(1.18kg)和水(0.65kg)装入搪玻璃反应器中。添加以上制备的化合物2的粗溶液(限量试剂),接着添加dipea(1.82kg)。将所得浆料在25℃下搅拌直至反应完成。通过过滤分离产物。用acn(1.6kg)洗涤粗固体。将滤饼在45℃下真空干燥。以98ap和83%的产率分离化合物3。步骤3:化合物8的制备在25℃下将水(6.0kg,6.0l/kg)和化合物7(1.0kg)装入搪玻璃反应器中。添加无水乙酸锌(1.08kg,1.0当量),接着添加化合物3(1.28kg,1.20当量)。用2-丙醇(0.79kg,1.0l/kg)和水(1.50kg,1.50l/kg)冲洗反应器管线。将所得的均匀溶液加热至65℃,并老化直至反应完成。添加水(7.0kg,7.0l/kg),并将粗混合物冷却至20℃并老化30min。通过过滤分离产物。依次用水(6.0kg,6.0l/kg)、水(6.0kg,6.0l/kg)、thf(5.3kg,6.0l/kg)和thf(5.3kg,6.0l/kg)洗涤粗固体。将滤饼在70℃下真空干燥。以98ap和94%的产率分离化合物8。步骤4:化合物9的制备用氮气吹扫另外的搪玻璃反应器。装入甲苯(0.87kg,1.0l/kg)和mecn(0.79kg,1.0l/kg),接着装入(2r)-1-[(1r)-1-[双(1,1-二甲基乙基)膦基]乙基]-2-(二环己基膦基)二茂铁(josiphossl-009-01)(14.1g,1.0mol%)和乙酸钯(2.9g,0.5mol%)。用甲苯(0.43kg,0.5l/kg)冲洗反应器管线。将所得的预形成的催化剂溶液保持在氮气下直到进一步使用。在20℃下,将甲苯(3.46kg,4.0l/kg)和acn(1.57kg,2.0l/kg)装入用氮气吹扫的搪玻璃反应器中。添加化合物8(1.00kg),接着添加dbu(0.39kg,1.00当量)。用甲苯(0.43kg,0.5l/kg)冲洗反应器管线。将化合物10(0.54kg,2.5当量)和k2co3(325目级,0.70kg,2.0当量)添加至反应混合物中,接着添加甲苯(1.30kg,1.5l/kg)和acn(0.79kg,1.0l/kg)。将预形成的催化剂溶液转移至反应混合物中,然后将其加热至75℃并搅拌直至反应完成。将反应粗品冷却至20℃。在1小时内缓慢装入乙酸水溶液(50体积%,4.0kg,4.0l/kg)。然后添加冰乙酸(10.5kg,10.0l/kg)。将所得的均匀溶液用庚烷(2×3.42kg,2×5.0l/kg)洗涤两次。收集底部水层,并转移至干净的反应器中。添加水(5.0kg,5.0l/kg),接着添加化合物9种子(0.01kg,1.0wt%)。将浆料在20℃下老化2小时。添加另外的水(2.0kg,2.0l/kg),并将浆料进一步老化6小时。通过过滤分离产物。将粗滤饼用acn水溶液(50体积%,4.5kg,5.0l/kg)洗涤,接着用acn(3.9kg,5.0l/kg)洗涤。将滤饼在65℃下真空干燥。以98.5ap和84%的产率分离化合物9。步骤5:化合物(i)的制备将nmp(2.06kg,2.0l/kg)和acn(0.78kg,1.0l/kg)装入搪玻璃反应器中,并在20℃下搅拌。将n-甲基咪唑(0.13kg,0.7当量)、化合物13(0.17kg,1.2当量)和化合物9(1.00kg)装入反应混合物中。将混合物加热至65℃并老化直至均匀。然后将20%湿润的hobt(0.17kg,0.5eq)、接着是edchcl(0.54kg,1.4eq)装入反应混合物中。用acn(0.78kg,1.0l/kg)冲洗反应器,然后将所得混合物在65℃下老化直至反应完成。通过装入水(1.0kg,1l/kg)猝灭反应,然后用acn(3.0kg,3l/kg)稀释。在冷却至0℃之前,将反应混合物在65℃下老化1h,并在0℃下再老化12h。通过过滤分离产物。将湿滤饼用2:1水:acn(2.8kg,3l/kg)洗涤,然后用acn(2.4kg,3l/kg)洗涤,然后在65℃下全真空下干燥。以>99.5%纯度和91%产率分离化合物(i)的纯度。将nmp(6.2kg,6.0l/kg)和化合物(i)(1.0kg)装入搪玻璃反应器中。将批料加热至70℃以形成浅黄色溶液,然后将其通过精制过滤器转移至70℃下的清洁容器中。添加2-丙醇(2.4kg,3l/kg),接着添加化合物(i)的种子(0.005kg,0.005kg/kg)。老化1小时后,在2小时的过程内装入(3l/kg/h)另外的2-丙醇(4.8kg,6l/kg)。将浆料在70℃下老化1h,缓慢冷却至0℃,然后在0℃下再老化12h。通过过滤分离产物。将湿滤饼用2-丙醇(2×3.1kg,2×4l/kg)洗涤,然后在65℃下在全真空下干燥。以>99.9%纯度和83%产率分离化合物(i)。化合物7的制备步骤1:n-甲基-n-甲酰肼的制备在0℃下向搪玻璃反应器中装入甲醇(1.6kg/kg,2.0l/kg)和甲基肼(1kg)。逐滴添加甲酸甲酯(0.57kg/kg,1.1当量)。将粗品加热至20℃并再老化6h。在真空下蒸馏粗品,直到总体积变为约0.5l/kg。用2-methf(5×3.6kg/kg)进行五次进/走(put/take)蒸馏以进行共沸干燥。将粗品冷却至20℃。将n-甲基-n-甲酰肼分离为89wt%-90wt%溶液,产率为89%-91%。步骤2:化合物5的制备在0℃下向搪玻璃反应器中装入叔丁醇钾(1.5kg/kg,2.4当量)和thf(12.2kg/kg)。缓慢添加化合物4(1.0kg)、n-甲基-n-甲酰肼(1.0kg/kg,2.30当量)和thf(5.3kg/kg,6.0l/kg)的混合物。用thf(0.5kg/kg)冲洗反应器管线。将反应粗品在0℃下老化直至反应完成。添加水(5.0kg/kg),并将所得混合物在0℃下老化30分钟,加热至40℃并再老化30min。分离各层,并丢弃水层。将有机层用盐水(15wt%,5.7kg/kg)洗涤,然后在真空下蒸馏直至总体积变为约5l/kg。用乙酸乙酯(4×10l/kg)进行四次进/走蒸馏以达到共沸干燥的目的。将粗品冷却至20℃。添加硫酸(0.66kg/kg,1.10当量),并将浆料搅拌2-3小时。通过过滤分离产物。将滤饼依次用乙酸乙酯(2×6.5l/kg)和庚烷(8l/kg)洗涤,并在45°℃下真空干燥。以99ap和83%的产率分离化合物5。步骤3:化合物6的制备在0-5℃下向搪玻璃反应器中装入浓硫酸(4.5kg/kg)和化合物5(1.0kg)。逐滴添加硝酸(68wt%,0.35kg/kg,1.2当量)。将混合物在0-5℃下搅拌直至反应完成。在单独的反应器中,将水(12kg/kg)和甲醇(6.5kg/kg,8.3l/kg)在20℃下充分混合。将硝化粗品缓慢转移到甲醇水混合物中。用甲醇(0.5kg/kg)冲洗反应器管线。将粗品加热至40℃-45℃。缓慢添加氢氧化铵水溶液(25wt%,7.4kg/kg)。将得到的浆料冷却至20℃并搅拌3h。通过过滤分离产物。将滤饼用水(2×6l/kg)洗涤,并在45℃下真空干燥。以99ap和95%的产率分离化合物6。步骤4:化合物7的制备向用氮气吹扫的高压反应器中装入甲醇(8.0kg/kg)和化合物6(1.0kg)。在仔细排除氧气的情况下,添加碳酸氢钠(0.6kg/kg,2.0当量)和pd/c(10%负载,50%湿润,0.02kg/kg)。用氢气(41-46psi)对反应器加压,并将反应混合物在20℃下老化6h,然后加热至45℃,并老化直至反应完成。用氮气吹扫反应器,并将反应粗品过滤以去除pd/c。使用甲醇(5kg/kg)帮助转移。在真空下蒸馏合并的滤液,直到总体积变为约2.5l/kg。添加水(10kg/kg),并将粗品在真空下蒸馏直至总体积变为约2.5l/kg。将粗品加热至70℃。添加盐水(25wt%,9.0kg/kg),并将所得的粗品在70℃下搅拌6h。冷却至0℃后,将粗品进一步老化6h。通过过滤分离产物。将滤饼用盐水洗涤(预冷至0℃,25wt%,2.0kg/kg),并在45°℃下真空干燥。以99ap和88%的产率分离化合物7。化合物13的制备步骤1:化合物11和化合物12的制备向用氮气吹扫的搪玻璃反应器中装入水(16.3l/kg)和氢氧化钠(3.3kg,3.0当量)。老化混合物直到氢氧化钠达到完全溶解。将粗品冷却至0℃。装入d4-甲醇(1.0kg)和thf(4.5l/kg)。在2h的过程内添加tscl(6.3kg,1.2当量)在thf(6.3kg,7.1l/kg)中的溶液。将粗品在0℃下搅拌直至反应完成。将批料加热至20℃。分离各层。将收集的有机层用mtbe(4.0kg,5.4l/kg)稀释,用盐水洗涤两次(25wt%,4.0kg,接着是12kg)。在真空下蒸馏有机层,直到总体积变为约10l/kg。用acn(2×10l/kg)进行两次进/走蒸馏以进行共沸干燥。将粗品冷却至20℃。添加acn(10.0kg,12.8l/kg)和nan(cho)2(3.3kg,1.2当量)。将粗品加热至65℃并搅拌直至反应完成。冷却至5℃后,将混合物过滤,并将粗滤饼用acn洗涤两次(2×2.5kg,2×3.2l/kg)。在真空下蒸馏合并的滤液,直到总体积变为约3l/kg。将粗品冷却至20℃。以80wt%-85wt%分离呈油的化合物12,产率为60%-70%。步骤2:实施例13的制备在20℃下向搪玻璃反应器中装入化合物12(1.0kg)和甲醇(3.9kg,5.0l/kg)。添加hcl在ipa中的溶液(5-6标准溶液,4.5kg,1.5当量)。将所得混合物加热至50℃并搅拌直至反应完成。缓慢添加thf(10kg,11.2l/kg),并在2h内将粗品冷却至0℃,以提供浆料。通过过滤分离产物。将滤饼用thf(3.7kg,4.1l/kg)洗涤,并在45℃下真空干燥。以80%产率分离化合物13为。化合物13的可选重结晶:将甲醇(5.6kg,8.3l/kg)和化合物13(1.0kg)装入搪玻璃反应器中。缓慢添加dbu(0.1kg)。将粗品搅拌1h。缓慢添加thf(12.4kg,13.9l/kg),并将所得的浆料老化2h。通过过滤分离产物。将滤饼用thf(2.6kg,2.9l/kg)洗涤,并在45℃下真空干燥。以60%产率分离化合物13(第1批)。在真空下蒸馏母液,直到总体积变为约1l/kg。用甲醇(2×2.8kg,2×3.6l/kg)进行两次进/走蒸馏,并将溶液浓缩回约1l/kg。将粗品冷却至20℃。添加thf(4.8kg,5.4l/kg),并将所得浆料老化2h。通过过滤分离产物。将滤饼用thf(1.0kg)洗涤,并在45℃下真空干燥。以25%产率分离化合物13(第2批)。实施例5:化合物(i)的形式b的物理和化学稳定性将化合物(i)的形式b的样品在不同的温度和湿度条件下储存4周。在第2周和第4周测量了以dsc、tga和pxrd为特征的物理稳定性。在至少4周内未在样品中检测到物理形式的变化。表3中的数据显示样品的化学稳定性没有任何测量变化。表4中的数据显示了形式b的长期稳定性。所示的%包括针对水和总挥发物校正的值,在一些情况下所述校正导致值大于100%。表3.表4.化合物(i)的形式b的吸湿等温线如图4所示。化合物(i)的形式b展示出在25℃下在25%与75%之间的相对湿度时吸湿率低于0.5%。表5显示了潜在盐的微溶出比较研究的结果。由于游离碱显示出ph效应,因此进行了该研究。显示出hcl盐比msa和硫酸盐(so4)更好地减轻ph效应。表5表6显示了dogpk的研究,其中测试了1mpk剂量下的游离碱相比于hcl盐的ph效应。在法莫替丁治疗的狗中,hcl盐的平均生物利用度比游离碱高60%。这与微溶出ph传递模型一致,所述模型显示与游离碱相比,hcl盐具有较低的ph效应风险。表6结果显示,化合物(i)的结晶形式b是不吸湿的材料,保留了良好的效能,没有明显的物理或化学降解,并且其杂质分布在测试的温度和湿度条件下保持不变。此外,相比于游离碱或其他盐形式,它减轻ph效应。单晶数据使用配备有铜阳极微焦点密封x射线管(cukα)和dectrispilatus3r200k混合像素阵列检测器的rigakusupernova衍射仪收集单晶x射线数据。单晶在数据收集过程中处于室温(约25℃)下。最终的晶胞参数是使用在4.1400°<θ<77.1740°范围内的4529反射的设置角度通过最小二乘法精修获得的。使用shelxt软件通过直接方法解析结构,并使用shelxl-2014软件通过全矩阵最小二乘法对结构进行精修(sheldrick,g.m.actacryst.2015,a71,3–8.;sheldrick,g.m.actacryst.,2008,a64,112–122.)。结构精修涉及最小化∑w(|fo|-|fc|)2定义的函数,其中w是基于观察到的强度误差的合适加权因子,fo是基于测得的反射的结构因子,并且fc是基于计算的反射的结构因子。通过使用残差因子r=∑||fo|-|fc||/∑|fo|和wr=[∑w(|fo|-|fc|)2/∑w|fo|]1/2评估精修的晶体结构模型与实验x射线衍射数据之间的一致性。在精修的所有阶段都检查了差分傅立叶图。使用各向异性热位移参数精修所有非氢原子。氢原子被独立地精修。pxrd使用具有vantec-500检测器的brukerc2gadds获得粉末x射线衍射(pxrd)数据。辐射为cukα(40kv,40ma)。样品检测器的距离为约20cm。入射光学器件包括goebel镜和0.3mm准直仪。将粉末样品置于直径为1mm或更小的密封玻璃毛细管中;在数据收集过程中旋转毛细管。针对2≤2θ≤35°收集数据,其中样品暴露时间为至少1000秒。将得到的二维衍射弧积分,以在约2至约30度2θ范围内建立了传统的步长为0.05度2θ的1维pxrd图。dsc差示扫描量热法(dsc)实验是在tainstrumentstmq2000型仪器上进行的。在铝盘中称量样品(约2-6mg),并准确记录至百分之一毫克,然后转移至dsc。用50ml/min的氮气吹扫仪器。在室温与300℃之间以10℃/min的加热速率收集数据。绘制吸热峰朝下的图。tga热重分析(tga)实验是在tainstrumentstmq5000型仪器上进行的。将样品(约10-30mg)置于先前加热过的铂盘中。准确地测量样品的重量,并用仪器记录至千分之一毫克。用氮气以100ml/min吹扫炉子。在室温与300℃之间以10℃/min的加热速率收集数据。吸湿等温线在tainstrumentvti-sa+vaporsorptionanalyzer中使用约10mg样品收集吸湿等温线。将样品在60℃下干燥直到获得0.0005wt%/min的损失率持续10分钟。在25℃下以及3%或4%、5%、15%、25%、35%、45%、50%、65%、75%、85%和95%rh下测试样品。当达到0.0003wt%/min的速率持续35分钟或最多600分钟时,达到每个rh的平衡。ssnmr在以400.13mhz的质子频率运行的brukeraviii仪器上进行了碳-13交叉极化魔角旋转(cpmas)固态nmr(ssnmr)实验。固体样品在4mmzro2转子中以13khz旋转。接触时间为2毫秒,并且在质子通道上从50%上升到100%(a.e.bennett等人,j.chem.phys.,1995,103,6951,g.metz,x.wu和s.o.smith,j.magn.reson.a.,1994,110,219-227)。在5×1ht1的api下保持弛豫延迟,即为16秒。使用带有3.8微秒脉冲(66khz标称带宽)的tppm序列进行质子去耦。光谱扫描宽度为300ppm,以100ppm为中心。采集2972个数据点(给出20hz的数字分辨率),并在切趾之前以20hz的谱线增宽从零填充至8192。共添加了3072个自由感应衰减。使用3-甲基戊二酸将光谱间接参照tms(d.barich,e.gorman,m.zell和e.munson,solidstatenuc.mag.res.,2006,30,125-129)。每个实验使用约70mg样品。温度设定为280°k。当前第1页1 2 3
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签:

 热门咨询
热门咨询




tips