
 商标分类
商标分类  商标转让
商标转让 
一种含烷基链的咪唑类磁性离子液体及其制备方法和应用与流程
 2021-02-02 17:02:14|
2021-02-02 17:02:14| 443|
443| 起点商标网
起点商标网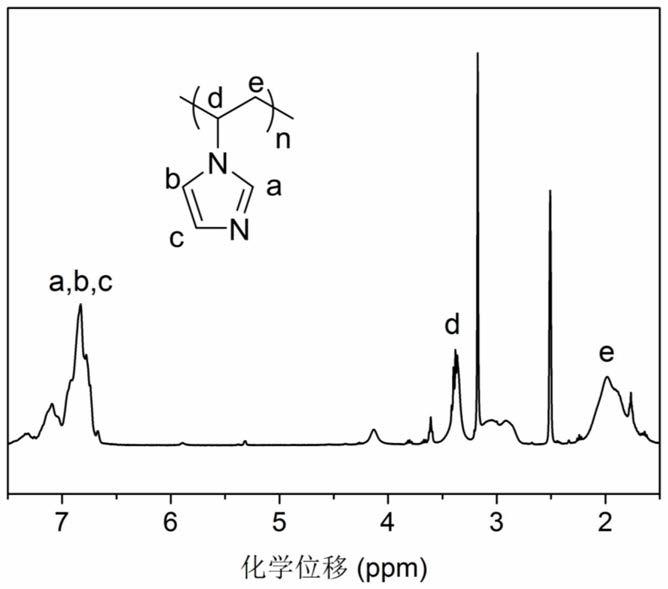
[0001]
本发明涉及聚离子液体技术领域,特别是涉及一种含烷基链的咪唑类磁性离子液体及其制备方法和应用。
背景技术:
[0002]
核磁共振成像技术(mri)已经成为了一种常用的非介入性的医学诊断手段。由于其具有良好的时空分辨率、软组织对比度、以及非电离辐射性能,该技术被广泛用于肿瘤、癌症等许多疾病的诊断。而通常来说,为了提高图像质量,需要使用mri造影剂(ca)来调节患病组织与正常组织之间的对比度。常用的造影剂通常是纵向弛豫时间造影剂(t
1
制剂),如含有镧系元素钆的络合物(l.tei et al.,dalton transactions,2009,9712-9714)。
[0003]
聚离子液体(pils)是一种在重复单元中包含一个可聚合的骨架以及一种离子液体的聚合物电解质。它既具有小分子离子液体的饱和蒸气压低、热稳定性、阻燃性以及高的离子导电性;又可以通过高分子的骨架自组装成多种多样的形态结构。通常,通过将pils的负离子交换为磁性阴离子络合物来对聚离子液体的磁性进行调控(z.xia et al.,polymer,2018,157,32-37)。
技术实现要素:
[0004]
本发明的目的是针对现有技术中存在的技术缺陷,而提供一种含烷基链的咪唑类磁性离子液体及其制备方法和在核磁成像中的应用。通过离子液体结构的引入,使得含烷基链的咪唑聚合物具备了良好的水溶性。另外将磁性负离子作为抗衡离子,使得其同时具备核磁成像的能力。实验结果证明,该离子液体可以用作极低浓度下的核磁成像造影剂。
[0005]
为实现本发明的目的所采用的技术方案是:
[0006]
一种含烷基链的咪唑类磁性离子液体,具有化学结构式a的结构且表现出磁性能;
[0007][0008]
其中,x表示烷基链中亚甲基的个数,x=0-19;n表示聚合物的聚合度,n=100-1000,x,n为整数。
[0009]
在上述技术方案中,x=1-11,n=300-500。
[0010]
上述含烷基链的咪唑类磁性离子液体的制备方法,包括以下步骤:
[0011]
s1:n-乙烯基咪唑与偶氮二异丁腈和4-氰基戊酸二硫代苯甲酸加热反应生成中间产物a;
[0012][0013]
s2:将s1得到的中间产物a与溴代烷在有机溶剂中加热反应,得到中间产物b;
[0014][0015]
所述溴代烷中,x为烷基链中亚甲基的个数,x=0-19,优选1-11;
[0016]
s3:向s2得到的中间产物b的甲醇溶液中滴加硝酸银水溶液室温反应以得到中间产物c;
[0017][0018]
s4:将s3得到的中间产物c与六水合硝酸钆,加热反应得到负离子为gd(no
3
)
4-的聚(乙烯基咪唑鎓盐)a。
[0019][0020]
在上述制备方法中,
[0021]
在s1中,偶氮二异丁腈、4-氰基戊酸二硫代苯甲酸和n-乙烯基咪唑的摩尔比为1:(5-20):(50-200);反应温度为40-100℃,优选50-80℃;反应时间6-36h,优选12-24h;
[0022]
在s2中,以甲醇做溶剂,反应温度为40-100℃,优选40-60℃,反应时间6-36h,优选
12-24h;
[0023]
在s3中,反应时间为10-60min,滴加速度为每分钟0.1—0.5ml,所述中间产物b和硝酸银的摩尔比为1:(2-5);
[0024]
在s4中,以乙腈做溶剂,反应温度为50-100℃,优选70-90℃,反应时间为12-36h,优选20-30h,所述中间产物c和六水合硝酸钆的摩尔比为1:(2-5)。
[0025]
在上述制备方法中,
[0026]
在s1中,反应完毕后使用大量乙醚洗涤产物,得到中间产物a;
[0027]
在s2中,反应完毕后使用大量乙醚洗涤产物,得到中间产物b;
[0028]
在s3中,反应结束后将产物过滤,滤液干燥后,得到中间产物c;
[0029]
在s4中,反应完毕后使用大量乙醚洗涤产物,得到负离子为gd(no
3
)
4-的聚(乙烯基咪唑鎓盐)a。
[0030]
上述含烷基链的咪唑类磁性离子液体的另一种制备方法,包括以下步骤:
[0031]
步骤1:将n-乙烯基咪唑与溴代烷在有机溶剂中加热反应,得到中间产物e,
[0032][0033]
所述溴代烷中,x为烷基链中亚甲基的个数,x=0-19,优选1-11;
[0034]
步骤2:将步骤1中得到的中间产物e,以n,n-二甲基甲酰胺为溶剂,4-氰基戊酸二硫代苯甲酸为链转移剂,偶氮二异丁腈为引发剂,加热反应生成中间产物b;
[0035][0036]
步骤3:向步骤2得到的中间产物b的甲醇溶液中滴加硝酸银水溶液室温反应以得到中间产物c;
[0037][0038]
步骤4:将步骤3得到的中间产物c与六水合硝酸钆,加热反应得到负离子为gd(no
3
)
4-的聚(乙烯基咪唑鎓盐)a。
[0039][0040]
在上述制备方法中,
[0041]
在步骤1中,以四氢呋喃做溶剂,反应温度为40-100℃,优选40-60℃,反应时间6-36h,优选12-24h,n-乙烯基咪唑和溴代烷的摩尔比为1:(2-10),优选1:(2-5);
[0042]
在步骤2中,偶氮二异丁腈、4-氰基戊酸二硫代苯甲酸和中间产物e的摩尔比为1:(5-20):(50-200);反应温度为40-100℃,优选50-80℃;反应时间6-36h,优选12-24h;
[0043]
在步骤3中,反应时间为10-60min,滴加速度为每分钟0.1—0.5ml,所述中间产物b和硝酸银的摩尔比为1:(2-5);
[0044]
在步骤4中,以乙腈做溶剂,反应温度为50-100℃,优选70-90℃,反应时间为12-36h,优选20-30h,所述中间产物c和六水合硝酸钆的摩尔比为1:(2-5)。
[0045]
上述含烷基链的咪唑类磁性离子液体,所述含烷基链的咪唑类磁性离子液体的水溶液的弛豫率范围为1-15mm-1
s-1
。
[0046]
本发明的另一方面,上述含烷基链的咪唑类磁性离子液体在核磁成像方面的应用。
[0047]
在上述应用中,体外核磁成像时,图像的亮度随离子液体水溶液的浓度增加而提高。
[0048]
与现有技术相比,本发明的有益效果是:
[0049]
本发明的技术方案中制备了结构可控的磁性聚(乙烯基咪唑鎓盐)聚离子液体。该化合物制备简单,具有良好的水溶性。另外该化合物在水中与水中的质子具有较强的相互作用,因而具备良好的成像效果。总之,本发明技术方案提供了一种新型、可行、高效的磁性聚离子液体合成方法,该方法反应条件温和、方法简单、原子利用率高,可以用做核磁成像的造影剂,在核磁成像方面具有巨大的应用前景。
附图说明
[0050]
图1所示为中间产物a的核磁共振谱图。
[0051]
图2所示为中间产物b1的核磁共振谱图。
[0052]
图3所示为中间产物c的核磁共振谱图。
[0053]
图4所示为中间产物e的核磁共振谱图。
[0054]
图5所示为离子液体a的弛豫率测试曲线。
[0055]
图6所示为离子液体a的体外核磁成像实验照片。
具体实施方式
[0056]
以下结合具体实施例对本发明作进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0057]
本发明中所用核磁共振谱仪为bruker avance iii 400mhz spectrometer;核磁共振成像研究系统为ht-mrsi60-60ky(60mm)。
[0058]
实施例1
[0059]
中间产物a的合成:
[0060][0061]
称取n-乙烯基咪唑10g,0.1mmol溶于10ml的二氧六环中,然后迅速向溶液中加入偶氮二异丁腈(0.2mmol,32.5mg)和4-氰基戊酸二硫代苯甲酸(1mmol,279.37mg),将反应充氮气-除氧气,冻融循环三次,然后在70℃的条件下反应24h。反应完毕,将产物用大量乙醚沉淀,离心,真空干燥24h后,得到淡黄色固体产物。收率:40%。图1为a的核磁共振谱图(
1
h-nmr,400hz,dmso-d
6
)。
[0062]
1
h nmr(400mhz,dmso-d
6
)δ:7.48
–
6.62(宽峰,咪唑环上的氢),3.55
–
3.25(宽峰,主链上次甲基上的氢),2.22
–
1.58(宽峰,主链上亚甲基上的氢)。
[0063]
中间产物b1的合成:
[0064][0065]
称取中间产物a(1g)溶于10ml甲醇中,并向溶液中加入1-溴丁烷(20mmol,2.7440g)。将反应加热至回流并保持回流反应36h,反应温度为70摄氏度。反应完毕,将反应冷却至室温,并用乙醚多次沉淀洗涤产物。真空干燥24h后,得到淡黄色固体产物。产率:100%。图2为b1的核磁共振谱图(
1
h-nmr,400hz,dmso-d
6
)。
[0066]
1
h nmr(400mhz,dmso-d
6
)δ:10.12
–
9.29(宽峰,咪唑环上两个氮原子之间的一个氢),8.49
–
8.55(宽峰,咪唑环上两个氮原子之间的两个氢),5.01
–
3.72(宽峰,主链上次甲
基上的氢以及与咪唑环相连的烷基链上的第一个亚甲基的氢),3.53
–
3.21(宽峰,主链上亚甲基上的氢),2.11
–
1.55(宽峰,与咪唑环相连的烷基链上的第二个亚甲基的氢),1.49
–
1.12(宽峰,与咪唑环相连的烷基链上的第三个亚甲基上的氢),1.05
–
0.70(宽峰,烷基链中甲基上的氢)。
[0067]
中间产物b2的合成:
[0068][0069]
称取中间产物a(1g)溶于10ml甲醇中,并向溶液中加入1-溴己烷(20mmol,3.3016g)。将反应加热至回流并保持回流反应36h,反应温度为70摄氏度。反应完毕,将反应冷却至室温,并用乙醚多次沉淀洗涤产物。真空干燥24h后,得到淡黄色固体产物。产率:100%。
[0070]
中间产物b3的合成:
[0071][0072]
称取中间产物a(1g)溶于10ml甲醇中,并向溶液中加入1-溴辛烷(20mmol,3.8626g)。将反应加热至回流并保持回流反应36h,反应温度为70摄氏度。反应完毕,将反应冷却至室温,并用乙醚多次沉淀洗涤产物。真空干燥24h后,得到淡黄色固体产物。产率:100%。
[0073]
中间产物b4的合成:
[0074][0075]
称取中间产物a(1g)溶于10ml甲醇中,并向溶液中加入1-溴癸烷(20mmol,4.4236g)。将反应加热至回流并保持回流反应36h,反应温度为70摄氏度。反应完毕,将反应冷却至室温,并用乙醚多次沉淀洗涤产物。真空干燥24h后,得到淡黄色固体产物。产率:100%。
[0076]
中间产物b5的合成:
[0077]
[0078]
称取中间产物a(1g)溶于10ml甲醇中,并向溶液中加入1-溴十二烷(20mmol,4.9846g)。将反应加热至回流并保持回流反应36h,反应温度为70摄氏度。反应完毕,将反应冷却至室温,并用乙醚多次沉淀洗涤产物。真空干燥24h后,得到淡黄色固体产物。产率:100%。
[0079]
中间产物c的合成:(后续制备方法中,以x=3为例,其余链长产物的制备方法与此方法一致)
[0080][0081]
称取中间产物b(0.1mmol,23.1mg),溶于5ml甲醇中(即均匀分散状态,全文同);称取agno
3
(0.2mmol,34.0mg)溶于0.5ml蒸馏水中配成溶液。在搅拌的条件(每分钟300转)下,将硝酸银水溶液滴入中间产物c溶液中进行反应(滴加速度为每分钟0.1ml),反应温度为室温20—25摄氏度,在滴加过程中两者反应已经开始,在滴加完毕后继续进行反应,共计反应1h。反应完毕,将反应所得混合物过滤,收集滤液,并将滤液在旋转蒸发仪上蒸发浓缩。将浓缩液在大量乙醚中沉淀3次,得到白色固体产物。产率100%。图3为c的核磁共振谱图(
1
h-nmr,400hz,dmso-d
6
)。
[0082]
1
h nmr(400mhz,dmso-d
6
)δ:10.12
–
9.29(宽峰,咪唑环上两个氮原子之间的一个氢),8.49
–
8.55(宽峰,咪唑环上两个氮原子之间的两个氢),5.01
–
3.72(宽峰,主链上次甲基上的氢以及与咪唑环相连的烷基链上的第一个亚甲基的氢),3.53
–
3.21(宽峰,主链上亚甲基上的氢),2.11
–
1.55(宽峰,与咪唑环相连的烷基链上的第二个亚甲基的氢),1.49
–
1.12(宽峰,与咪唑环相连的烷基链上的第三个亚甲基上的氢),1.05
–
0.70(宽峰,烷基链中甲基上的氢)。
[0083]
含烷基链的咪唑类磁性离子液体a的合成:
[0084][0085]
称取中间产物c(0.1mmol,21.3mg)溶于乙腈中;并加入六水硝酸钆(0.1mmol,45.1mg)在80℃下搅拌(每分钟200转)反应24h。反应完毕,将反应物在旋转蒸发仪上蒸发浓缩,并将浓缩液在大量乙醚中多次沉淀,真空干燥,得到白固体产物。产率100%。
[0086]
实施例2
[0087]
本实施例介绍另一种制备方法。
[0088]
中间产物e的合成:
[0089]
[0090]
称取n-乙烯基咪唑(53.13mmol,5g),溶于10ml四氢呋喃中,然后向溶液中加入1-溴丁烷(106.26mmol,14.56g)。将反应在60℃的条件下反应24h,冷凝回流。所得产物旋转蒸发浓缩,粗产物用乙醚多次沉淀,真空干燥后,得到淡黄色油状产物。图5为e的核磁共振谱图(
1
h-nmr,400hz,cdcl
3
)。
[0091]
1
h nmr(400mhz,cdcl
3
)δ:11.12(单重峰,咪唑环上两个氮原子之间的一个氢),7.76,7.51(单重峰,唑环上两个氮原子之间的两个氢以及乙烯基上与n相连的碳上的一个氢),5.93,5.43(多重峰,乙烯基远离n元素的碳上的两个氢),4.46(三重峰,与咪唑环上n相连的亚甲基上的两个氢),1.84(五重峰,季铵盐侧链上中间一个亚甲基上的两个氢),1.44(五重峰,季铵盐侧链上中间一个亚甲基上的两个氢),0.99(三重峰,季铵盐侧链上的甲基上的三个氢)
[0092]
中间产物b的合成:
[0093][0094]
称取中间产物e(11.557g)溶于10ml的n,n-二甲基甲酰胺中,然后迅速向溶液中加入偶氮二异丁腈(0.2mmol,32.5mg)和4-氰基戊酸二硫代苯甲酸(1mmol,279.37mg),将反应充氮气-除氧气,冻融循环三次,然后在70℃的条件下反应24h。反应完毕,将产物用大量乙醚沉淀,离心,真空干燥24h后,得到淡黄色固体产物。收率:40%。
[0095]
中间产物c的合成:
[0096][0097]
称取中间产物b(0.1mmol,23.1mg),溶于5ml甲醇中(即均匀分散状态,全文同);称取agno
3
(0.2mmol,34.0mg)溶于0.5ml蒸馏水中配成溶液。在搅拌的条件(每分钟300转)下,将硝酸银水溶液滴入中间产物c溶液中进行反应(滴加速度为每分钟0.1ml),反应温度为室温20—25摄氏度,在滴加过程中两者反应已经开始,在滴加完毕后继续进行反应,共计反应1h。反应完毕,将反应所得混合物过滤,收集滤液,并将滤液在旋转蒸发仪上蒸发浓缩。将浓缩液在大量乙醚中沉淀3次,得到白色固体产物。产率100%。
[0098]
含烷基链的咪唑类磁性离子液体a的合成:
[0099][0100]
称取中间产物c(0.1mmol,21.3mg)溶于乙腈中;并加入六水硝酸钆(0.1mmol,
45.1mg)在80℃下搅拌(每分钟200转)反应24h。反应完毕,将反应物在旋转蒸发仪上蒸发浓缩,并将浓缩液在大量乙醚中多次沉淀,真空干燥,得到白固体产物。产率100%。
[0101]
实施例3
[0102]
实施例1或实施例2制备的含烷基链的咪唑类磁性离子液体a的弛豫率测试实验,方法如下:
[0103]
将含烷基链的咪唑类磁性离子液体a配成浓度0-1mm的水溶液,装入1.5ml的离心管中,然后在核磁共振成像研究系统上进行测试,测试磁场强度为1.5t。得到的纵向弛豫时间(t
1
)。对于不同实验数据,做纵向弛豫时间倒数(1/t
1
)与浓度的关系曲线,如图5所示,其斜率为纵向弛豫率r
1
根据测试结果求得,弛豫率范围为1-15mm-1
s-1
。
[0104]
实施例4
[0105]
实施例1或实施例2制备的含烷基链的咪唑类磁性离子液体(a)的体外核磁成像实验,方法如下:将含烷基链的咪唑类磁性离子液体a配成浓度0-1mm的水溶液,装入1.5ml的离心管中,然后再核磁共振成像研究系统上进行体外成像测试,测试磁场强度为1.5t。图6为含烷基链的咪唑类磁性离子液体a的体外核磁成像实验照片。如图可知。随着样品浓度的增加,图像亮度也不断提高,说明成像效果不断提高,该含烷基链的咪唑类磁性离子液体a具备一定的核磁成像造影效果。
[0106]
依照本发明内容进行工艺参数调整,均可制备本发明的含烷基链的咪唑类磁性离子液体,并表现出与实施例1基本一致的性能。
[0107]
以上所述仅是本发明的优选实施方式,应当指出的是,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips