
 商标分类
商标分类  商标转让
商标转让 
一种乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体及其制备方法和应用与流程
 2021-02-02 16:02:16|
2021-02-02 16:02:16| 512|
512| 起点商标网
起点商标网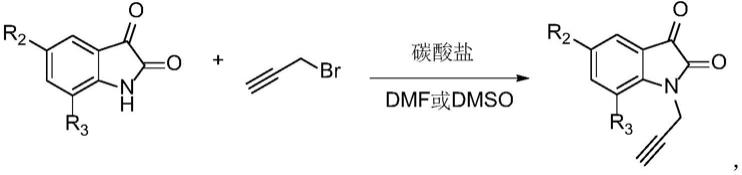
[0001]
本发明涉及医药化学技术领域,具体涉及一种乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体及其制备方法和应用。
背景技术:
[0002]
随着抗菌素的广泛、长期使用甚至滥用,细菌包括革兰阳性菌、阴性菌和其它致病菌对几乎所有的抗生素均产生了不同程度的耐药性
[1,2]
。耐药菌的不断涌现和在世界范围内的广泛传播不仅已成为临床医师所必需经常面对的棘手问题,而且因此丧命的患者人数不断攀升
[3,4]
。据估计,每年约有70万人死于与耐药菌相关的疾病,且这一数字还在持续增长
[5,6]
。因此,研发对药敏型和耐药型致病菌具有活性的新型抗生素迫在眉睫。
[0003]
首个喹诺酮
--
萘啶酸是1962年lesher等在合成抗疟疾药物氯喹时偶然发现的副产品,其对大肠埃希菌、克雷伯菌属、变形杆菌属、肠杆菌属等革兰阴性菌具有中等强度的活性
[7,8]
。萘啶酸于1964年用于临床治疗敏感革兰阴性菌所致的尿路感染,但因抗菌谱窄和易产生耐药性等缺陷目前已退出市场。
[0004]
自上世纪80年代初,koga等结合吡哌酸和氟甲喹的结构特征成功开发出6-氟-7-哌嗪基取代的诺氟沙星(首个氟喹诺酮、喹诺酮发展史上具有划时代意义的品种)以来,这类药物发展到了全新阶段
[9,10]
。研究证实,c-6位的氟原子可以增强此类化合物对dna促旋酶的抑制作用和提高对细胞膜的渗透性,进而增强抗菌活性,故目前临床上使用的喹诺酮类抗菌药绝大多数属于氟喹诺酮类
[11,12]
。氟喹诺酮类抗菌药通过与细菌dna促旋酶(水解三磷酸腺苷所必需的拓扑异构酶ii)或拓扑异构酶iv发生交互作用形成三元复合物,诱导dna促旋酶和拓扑异构酶iv发生构型改变,进而导致dna降解及菌体死亡。喹诺酮对革兰阳性菌的主要作用靶点为拓扑异构酶iv,而对革兰阳性菌的主要作用靶点则为dna促旋酶。细菌的促旋酶与哺乳动物差异明显,而喹诺酮对二者的选择性相差上千倍,故总体而言,喹诺酮的安全性良好。目前,氟喹诺酮在临床上主要用于治疗上、下呼吸道感染、胃肠道感染、妇科感染、性传播疾病、前列腺炎、骨关节感染、皮肤和软组织感染等
[13,14]
,引起了药物化学家的普遍关注。然而,与其他药物一样,细菌对喹诺酮类药物的耐药性也成逐年上升之势,已成为世界范围内的棘手问题
[15,16]
。为对付耐药菌,药物化学家近年来有针对性地合成了数以十万计的新喹诺酮类化合物,并取得了可喜成果,新品种不断问世。
[0005]
氟喹诺酮结构骨架可供修饰的位点较多,近年来药物化学家对其进行了系统而广泛的结构修饰
[17,18]
。其中,氟喹诺酮的c-7位取代基与抗菌谱、抗菌活性、药代动力学性质和安全性等密切相关,被认为是最适合修饰的位点
[14,15]
。最近,将其它活性分子或药效团通过适当的连接子揉合到氟喹诺酮c-7位的杂合体策略引起了科学家的广泛关注
[19,20]
。杂合体分子不仅可能具有双重或多重作用机制增强对耐药菌的活性,还具有拓展抗菌谱、改善药代动力学和减少毒副作用的潜质
[21,22]
。目前,多个氟喹诺酮杂合体如β-内酰胺-氟喹诺酮杂合体ro-23-9424、噁唑烷酮-氟喹诺酮杂合体mcb3681和利福霉素-氟喹诺酮杂合体
cbr-2092等对包括耐药菌在内的多种致病菌具有良好的活性,三者正处于临床评价阶段,有望于不久的将来为人类健康服务
[23,24]
。显然,将氟喹诺酮与其它具有抗菌活性的药效团杂合是获得对药敏型和耐药型致病菌均有效的新型抗菌药的有效途径。
[0006]
β-内酰胺-氟喹诺酮杂合体ro-23-9424、噁唑烷酮-氟喹诺酮杂合体mcb3681和利福霉素-氟喹诺酮杂合体cbr-2092的化学式如下:
[0007][0008]
靛红又称吲哚满二酮广泛存在于自然界中,具有包括抗菌活性在内的多种生物活性
[25,26]
。其中,某些靛红衍生物如司马沙尼、舒尼替尼和尼达尼布等目前已应用于临床或处于临床研究阶段
[27,28]
。因此,靛红也是药物化学领域常见的药效团。由于氟喹诺酮和靛红均具有潜在的抗菌活性,故氟喹诺酮-靛红杂合体是研发对药敏型和耐药型致病菌均有效的新型候选物的良好选择。事实上,多个氟喹诺酮-靛红杂合体显示出良好的抗菌活性,如对次丙基连接的靛红-环丙沙星杂合体的体外抗菌活性研究结果表明,这类杂合体对所测8株革兰阳性菌(mic:0.125-64μg/ml)和15株阴性菌(mic:≤0.03-2.0μg/ml)具有良好的活性
[29-38]
。其中,杂合体1对包括耐甲氧西林金葡球菌(mrsa)和耐甲氧西林表葡球菌(mrse)在内的多种耐药型和药敏型革兰阳性菌(mic:0.125-8μg/ml)和阴性菌(mic:≤0.03-0.5μg/ml)的活性与环丙沙星和左氧氟沙星相当或更优。对杂合体1(50mg/kg,口服)的代谢稳定性和体内药代动力学性质研究结果表明,该杂合体1可在1.2h达到最大血药浓度,最大血药浓度为832ng/ml,半衰期为3.3h,药时曲线下面积为2865ng
·
h/ml,具有深入研究价值。
[0009]
杂合体1的化学结构式为:
[0010]
构-效关系(sar)研究显示,氟喹诺酮与靛红之间的连接子对抗菌活性有显著影响
[39-48]
。而1,2,3-三氮唑不仅可经过点击化学方便制得,而且可通过形成氢键和范德华力等多种非共价键作用与作用位点相结合,进而增强生物活性,故1,2,3-三氮唑是良好的连
接子,在药物化学领域备受青睐
[49,50]
。
技术实现要素:
[0011]
基于上述现有技术存在的问题,本发明的目的是在于提供了一种乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体及其制备方法和应用,该杂合体对革兰氏阳性菌和阴性菌具有良好的活性,可作为抗菌药物候选物。
[0012]
该杂合体的制备方法简单,合成步骤少,合成条件易控制。
[0013]
实现本发明上述目的所采用的技术方案为:
[0014]
一种乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体,其结构通式为:
[0015][0016]
其中,r
1
为氧、羟亚胺、甲氧亚胺或乙氧亚胺,r
2
为氢、氟、氯或甲基,r
3
为氢、氟、氯或甲基。
[0017]
一种乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体的制备方法,包括如下步骤:
[0018]
1、在n,n-二甲基甲酰胺或二甲基亚砜中,在碳酸盐做碱的条件下,常温下将靛红及其衍生物与3-溴丙炔进行取代反应,得到n-炔丙基靛红及其衍生物,其反应方程式如下:
[0019][0020]
其中,r
2
为氢、氟、氯或甲基,r
3
为氢、氟、氯或甲基;
[0021]
2、在四氢呋喃和水的混合溶剂中,在碳酸氢钠存在的条件下,常温下将n-炔丙基靛红及其衍生物与盐酸羟胺或烷氧胺盐酸盐(r
4
onh
2
.hcl)进行反应,得到c-3位含有肟基的靛红中间体,其反应方程式如下:
[0022][0023]
其中,r
4
为甲基、乙基或氢;
[0024]
3、将四氢呋喃中,在二环己基碳二亚胺存在的条件下,将叠氮乙酸与n-羟基琥珀酰亚胺进行缩合反应,得到2-叠氮乙酸琥珀酰亚胺活性酯,其反应方程式如下:
[0025][0026]
4、在n,n-二甲基甲酰胺溶剂中,以n,n-二异丙基乙胺做碱,常温下将2-叠氮乙酸琥珀酰亚胺活性酯和加替沙星进行反应,得c-7位连有2-叠氮乙酰基的加替沙星衍生物,其反应方程式如下:
[0027][0028]
5、在n,n-二甲基甲酰胺中,以醋酸铜为催化剂,在30-70℃下将n-炔丙基靛红及其衍生物与2-叠氮乙酰基的加替沙星衍生物进行环化反应,生成乙酰基连接的环丙沙星-1,2,3-三氮唑-靛红杂合体,其反应方程式如下:
[0029][0030]
进一步,所述的碳酸盐为碳酸钾或碳酸钠。
[0031]
一种乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体在制备抗菌药物中的应用。
[0032]
与现有技术相比,本发明的有益效果和优点在于:
[0033]
该杂合体在大多数革兰阳性菌和阴性菌具有良好的活性,能作为抗菌药物的候选物,具有广阔的应用前景。
具体实施方式
[0034]
下面结合具体实施例对本发明进行详细说明。
[0035]
实施例1
[0036]
1、n-炔丙基靛红的合成:
[0037]
将3g(0.0205mol)靛红用50ml n,n-二甲基甲酰胺(dmf)溶解,再加入2.44g 3-溴
丙炔(0.0205mol)于250ml三颈烧瓶中反应,然后加入8.50g碳酸钾(0.0615mol),在常温下搅拌反应,tlc跟踪反应进程,10小时后反应完毕,向三颈烧瓶中加300ml水稀释,150ml乙酸乙酯萃取,用饱和食盐水洗涤,合并有机相,蒸干,得粗品,经硅胶柱层析纯化,得2.44g n-炔丙基靛红(0.0114mol);
[0038]
此步骤的反应方程式为:
[0039][0040]
2、2-叠氮乙酸琥珀酰亚胺活性酯的合成:
[0041]
将100mmol叠氮乙酸溶解在200ml四氢呋喃中,加入120.0mmol二环己基碳二亚胺和105.0mmol n-羟基琥珀酰亚胺,在室温下搅拌12h,过滤,滤液即2-叠氮乙酸琥珀酰亚胺活性酯的四氢呋喃溶液,无需纯化,直接使用;
[0042]
此步骤的反应方程式为:
[0043][0044]
3、c-7位连有2-叠氮乙酰基的加替沙星衍生物的合成:
[0045]
向100ml dmf中加入20mmol加替沙星、48ml含上述2-叠氮乙酸琥珀酰亚胺活性酯的四氢呋喃溶液和100ml dipea,于室温下搅拌24小时。除去溶剂后,残余物用硅胶柱提纯(洗脱剂dcm:meoh=10:1体积比)得c-7位连有2-叠氮乙酰基的加替沙星衍生物;
[0046]
此步骤的反应方程式为:
[0047][0048]
4、乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体的合成:
[0049]
向50ml dmf中加入10mmol 2-叠氮乙酰基的加替沙星衍生物、12mmol n-炔丙基靛红和1mmol醋酸铜cu(oac)
2
,在50℃下搅拌2h,然后在减压下浓缩,过反相柱,得到1-环丙基-7-(4-(2-(4-((靛红-1-基)甲基)-1h-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-6-氟-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率63%;
[0050]
此步骤的反应方程式为:
[0051]
[0052]
1h nmr(400mhz,dmso-d6)δ1.03-1.43(7h,m,环丙基-4h和-ch
3
),3.15-3.56(5h,m,哌嗪基-5h),3.74-4.26(6h,m,哌嗪基-2h,环丙基-1h和-och
3
),5.01(2h,s,-ch
2-连接基),5.53-5.65(2h,m,-ch
2-连接基),7.15(1h,t,j=8.0hz,ar-h),7.20(1h,d,j=8.0hz,ar-h),7.59(1h,d,j=8.0hz,ar-h),7.66(1h,t,j=8.0hz,ar-h),7.80(1h,d,j=8.0hz,ar-h),8.11(1h,s,ar-h),8.72(1h,s,ar-h),14.92(1h,brs,cooh)。
[0053]
hrms-esi:m/z c
32
h
31
fn
7
o
7
,[m+h]
+
的分析计算值:644.22635;实测:644.22407。
[0054]
实施例2
[0055]
与实施例1的操作步骤相同,只是将靛红换成5-氟靛红,5-氟靛红的结构式为:
[0056][0057]
最终得到1-环丙基-6-氟-7-(4-(2-(4-((5-氟靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率59%,其结构式为:
[0058][0059]
1h nmr(400mhz,dmso-d6)δ1.01-1.42(7h,m,环丙基-4h和-ch
3
),3.17-3.55(5h,m,哌嗪基-5h),3.73-4.26(6h,m,哌嗪基-2h,环丙基-1h和-och
3
),5.01(2h,s,-ch
2-连接基),5.43-5.65(2h,m,-ch
2-连接基),7.22(1h,d,j=4.0hz,ar-h),7.48-7.56(2h,m,ar-h),7.76(1h,d,j=12.0hz,ar-h),8.11(1h,s,ar-h),8.71(1h,s,ar-h),14.94(1h,brs,cooh)。
[0060]
hrms-esi:m/z c
32
h
30
f
2
n
7
o
7
,[m+h]+的计算值:662.21693;实测:662.21518。
[0061]
实施例3
[0062]
与实施例1的操作步骤相同,只是将靛红换成5-甲基靛红,5-甲基靛红的结构式为:
[0063][0064]
最终得到1-环丙基-6-氟-7-(3甲基-4-(2-(4-((5-甲基靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)哌嗪-1-基)-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率71%,其结构式为:
[0065][0066]
1h nmr(400mhz,cdcl
3
)δ1.01-1.52(7h,m,环丙基-4h和-ch
3
),2.32(3h,s,-ch
3
),3.26-3.55(6h,m,哌嗪基-6h),3.71-3.75(4h,m,哌嗪基-1h和-och
3
),4.01-4.02(1h,m,环丙基-1h),5.02(2h,s,-ch2-连接基),5.22-5.39(2h,m,-ch
2-连接基),7.16(1h,d,j=8.0hz,ar-h),7.38-7.40(2h,m,ar-h),7.83-7.86(2h,m,ar-h),8.82(1h,s,ar-h)。
[0067]
实施例4
[0068]
与实施例1的操作步骤相同,只是将靛红换成7-氟靛红,7-氟靛红的结构式:
[0069][0070]
最终得到1-环丙基-6-氟-7-(4-(2-(4-((7-氟靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率43%,其结构式为:
[0071][0072]
1h nmr(400mhz,cdcl
3
)δ1.01-1.38(7h,m,环丙基-4h和-ch
3
),3.30-3.57(4h,m,哌嗪基-4h),3.76(3h,s,-och
3
),4.03-4.04(1h,m,环丙基-1h),4.47-4.82(3h,m,哌嗪基-3h),5.23(2h,s,-ch
2-连接基),5.32-5.39(2h,m,-ch
2-连接基),7.12(1h,t,j=4.0hz,ar-h),7.34-7.39(1h,m,ar-h),7.44(1h,d,j=8.0hz,ar-h),7.87-7.92(2h,m,ar-h),8.82(1h,s,ar-h),14.82(1h,brs,cooh)。
[0073]
hrms-esi:m/z c
30
h
26
f
2
n
7
o
6
[m+h]+的计算值:618.19071;实测:618.18848。
[0074]
实施例5
[0075]
与实施例1的操作步骤相同,只是将靛红换成5,7-二氯靛红,5,7-二氯靛红的结构式:
[0076]
[0077]
最终得到1-环丙基-7-(4-(2-(4-((5,7-二氯靛红-1-基)甲基)-1h-1,2,3-三唑-1-基)乙酰基-3-甲基哌嗪-1-基)-6-氟-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率22%,其结构式为:
[0078][0079]
1h nmr(400mhz,dmso-d6)δ1.02-1.42(7h,m,环丙基-4h和-ch
3
),3.14-3.55(6h,m,哌嗪基-6h),3.73-3.81(4h,m,哌嗪基-1h和-och
3
),4.15-4.16(1h,m,环丙基-1h),5.29(2h,s,-ch
2-连接基),5.43-5.69(2h,m,-ch
2-连接基),7.20(1h,d,j=8.0hz,ar-h),7.64(1h,d,j=12.0hz,ar-h),7.76(1h,d,j=12.0hz,ar-h),8.12(1h,s,ar-h),8.72(1h,s,ar-h),14.91(1h,brs,cooh)。
[0080]
hrms-esi:m/z c
32
h
29
cl
2
fn
7
o
7
[m+h]+计算值:712.14841;实测:712.14639。
[0081]
实施例6
[0082]
1、n-炔丙基靛红的合成:
[0083]
将3g(0.0205mol)靛红用50ml n,n-二甲基甲酰胺(dmf)溶解,再加入2.44g 3-溴丙炔(0.0205mol)于250ml三颈烧瓶中反应,然后加入8.50g碳酸钾(0.0615mol),在常温下搅拌反应,tlc跟踪反应进程,10小时后反应完毕,向三颈烧瓶中加300ml水稀释,150ml乙酸乙酯萃取,用饱和食盐水洗涤,合并有机相,蒸干,得粗品,经硅胶柱层析纯化得2.44gn-丙炔基靛红(0.0114mol);
[0084]
此步骤的反应方程式为:
[0085][0086]
2、n-丙炔基-3-甲氧亚胺基靛红的合成:
[0087]
将1.0mmol n-炔丙基靛红溶解在10ml四氢呋喃和水的混合溶液中,加入1.2mmol甲氧胺盐酸盐和3.0mol碳酸氢钠固体,在室温下搅拌12h,然后加200ml水进行稀释,接着用150ml乙酸乙酯进行萃取,再用饱和食盐水洗涤,合并有机相,蒸干,得粗品,经硅胶柱层析纯化,得n-丙炔基-3-甲氧亚胺基靛红;
[0088]
此步骤的反应方程式为:
[0089]
[0090]
3、2-叠氮乙酸琥珀酰亚胺活性酯的合成:
[0091]
将100mmol叠氮乙酸溶解在200ml四氢呋喃中,加入120.0mmol二环己基碳二亚胺和105.0mmol n-羟基琥珀酰亚胺,在室温下搅拌12h,过滤,滤液即2-叠氮乙酸琥珀酰亚胺活性酯的四氢呋喃溶液,无需纯化,直接使用;
[0092]
此步骤的反应方程式为:
[0093][0094]
4、c-7位连有2-叠氮乙酰基的加替沙星衍生物的合成:
[0095]
向100ml dmf中加入20mmol加替沙星、48ml上述2-叠氮乙酸琥珀酰亚胺活性酯的四氢呋喃溶液和100ml dipea,于室温下搅拌24小时。除去溶剂后,残余物用硅胶柱提纯(洗脱剂dcm:meoh=10:1体积比)得c-7位连有2-叠氮乙酰基的加替沙星衍生物;
[0096]
此步骤的反应方程式为:
[0097][0098]
5、乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体的合成:
[0099]
向50ml dmf中加入10mmol 2-叠氮乙酰基的加替沙星衍生物、12mmol n-炔丙基靛红和1mmol醋酸铜cu(oac)
2
,在50℃下搅拌2h,然后在减压下浓缩,过反相柱,得到1-环丙基-7-(4-(2-(4-((3-甲氧亚胺基靛红-1-基)甲基)-1h-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-6-氟-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率79%;其反应方程式为:
[0100][0101]
1hnmr(400mhz,cdcl
3
)δ1.01-1.49(7h,m,环丙基-4h和-ch
3
),3.29-3.55(5h,m,哌嗪基-5h),3.74-3.76(4h,m,哌嗪基-1h和-och
3
),4.02-4.03(1h,m,环丙基-1h),4.27-4.46(4h,m,哌嗪基-1h和-noch
3
),5.09(2h,s,-ch
2-连接基),5.19-5.32(2h,m,-ch
2-连接基),7.06(1h,t,j=8.0hz,ar-h),7.20(1h,d,j=8.0hz,ar-h),7.36(1h,d,j=8.0hz,ar-h),7.81(1h,d,j=8.0hz,ar-h),7.90-7.95(2h,m,ar-h),8.84(1h,s,ar-h)。
[0102]
hrms-esi:m/z c
33
h
34
fn
8
o
7
[m+h]+计算值:673.25290;实测:673.24989。
[0103]
实施例7
[0104]
与实施例6的操作步骤相同,只是将靛红换成5-氟靛红,得到1-环丙基-6-氟-7-(4-(2-(4-((5-氟-3-甲氧基亚胺基靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率68%,其结构式为:
[0105][0106]
1hnmr(400mhz,cdcl
3
)δ1.02-1.38(7h,m,环丙基-4h和-ch
3
),3.29-3.54(5h,m,哌嗪基-5h),3.73-3.77(4h,m,哌嗪基-1h和-och
3
),4.02-4.03(1h,m,环丙基-1h),4.28-4.32(4h,m,哌嗪基-1h和-noch
3
),5.08(2h,s,-ch
2-连接基),5.20-5.32(2h,m,-ch
2-连接基),7.08-7.20(2h,m,ar-h),7.68(1h,d,j=4.0hz,ar-h),7.82(1h,s,ar-h),7.92(1h,d,j=12.0hz,ar-h),8.85(1h,s,ar-h),14.65(1h,brs,cooh)。
[0107]
hrms-esi:m/z c
33
h
33
f
2
n
8
o
7
[m+h]+计算值:691.24348;实测:691.24071。
[0108]
实施例8
[0109]
与实施例6的操作步骤相同,只是将靛红换成7-氟靛红,得到1-环丙基-6-氟-7-(4-(2-(4-((7-氟-3-甲氧基亚胺基靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率57%,其结构式为:
[0110][0111]
1hnmr(400mhz,dmso-d6)δ0.82-1.43(7h,m,环丙基-4h和ch
3
),3.13-3.24(5h,m,哌嗪基-5h),3.70-3.79(4h,m,哌嗪基-1h和-och
3
),3.98-3.99(1h,m,环丙基-1h),4.19-4.26(4h,m,哌嗪基-1h和-noch
3
),5.09(2h,s,-ch
2-连接基),5.40-5.63(2h,m,-ch
2-连接基),7.10-7.15(1h,m,ar-h),7.38(1h,t,j=8.0hz,ar-h),7.66(1h,d,j=12.0hz,ar-h),7.78(1h,t,j=8.0hz,ar-h),8.02(1h,s,ar-h),8.64(1h,s,ar-h)。
[0112]
hrms-esi:m/z c
33
h
33
f
2
n
8
o
7
[m+h]+计算值:691.24348;实测:691.24052。
[0113]
实施例9
[0114]
1、n-丙炔基靛红的合成:
[0115]
将3g(0.0205mol)靛红用50ml n,n-二甲基甲酰胺(dmf)溶解,再加入2.44g 3-溴丙炔(0.0205mol)于250ml三颈烧瓶中反应,然后加入8.50g碳酸钾(0.0615mol),在常温下搅拌反应,tlc跟踪反应进程,10小时后反应完毕,向三颈烧瓶中加300ml水稀释,150ml乙酸
乙酯萃取,用饱和食盐水洗涤,合并有机相,蒸干,得粗品,经硅胶柱层析纯化得2.44g n-丙基炔靛红(0.0114mol);
[0116]
此步骤的反应方程式为:
[0117][0118]
2、n-炔丙基-3-羟亚胺基靛红的合成:
[0119]
将1.0mmol n-炔丙基靛红溶解在10ml四氢呋喃和水的混合溶液中,加入1.2mmol羟胺和3.0mol碳酸氢钠固体,在室温下搅拌12h,然后加20ml水进行稀释,接着用150ml乙酸乙酯进行萃取,再用饱和食盐水洗涤,合并有机相,蒸干,得粗品,经硅胶柱层析纯化,得n-丙炔基-3-羟亚胺基靛红;
[0120]
此步骤的反应方程式为:
[0121][0122]
3、2-叠氮乙酸琥珀酰亚胺活性酯的合成:
[0123]
将100mmol叠氮乙酸溶解在200ml四氢呋喃中,加入120.0mmol二环己基碳二亚胺和105.0mmol n-羟基琥珀酰亚胺,在室温下搅拌12h,过滤,滤液即2-叠氮乙酸琥珀酰亚胺活性酯的四氢呋喃溶液,无需纯化,直接使用;
[0124]
此步骤的反应方程式为:
[0125][0126]
4、c-7位连有2-叠氮乙酰基的加替沙星衍生物的合成:
[0127]
向100ml dmf中加入20mmol加替沙星、48ml上述2-叠氮乙酸琥珀酰亚胺活性酯的四氢呋喃溶液和100ml dipea,于室温下搅拌24小时。除去溶剂后,残余物用硅胶柱提纯(洗脱剂dcm:meoh=10:1体积比)得c-7位连有2-叠氮乙酰基的加替沙星衍生物;
[0128]
此步骤的反应方程式为:
[0129]
[0130]
5、乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体的合成:
[0131]
向50ml dmf中加入10mmol 2-叠氮乙酰基的加替沙星衍生物、12mmol n-炔丙基-3-羟亚胺基靛红和1mmol cu(oac)
2
,在50℃下搅拌2h,然后在减压下浓缩,过反相柱,得1-环丙基-6-氟-7-(4-(2-(4-((3-羟亚胺基靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率:47%,其反应方程式为:
[0132][0133]
1hnmr(400mhz,dmso-d6)δ1.00-1.42(7h,m,环丙基-4h和ch
3
),3.16-3.55(5h,m,哌嗪基-5h),3.73-3.81(4h,m,哌嗪基-1h和-och
3
),4.15-4.25(2h,m,哌嗪基-1h和环丙基-1h),5.03(2h,s,-ch
2-连接基),5.76(2h,s,-ch
2-连接基),7.09(1h,t,j=8.0hz,ar-h),7.18(1h,d,j=8.0hz,ar-h),7.42(1h,t,j=8.0hz,ar-h),7.76(1h,d,j=12.0hz,ar-h),8.02(1h,s,ar-h),8.32(1h,s,ar-h),8.74(1h,s,ar-h)。
[0134]
hrms-esi:m/z c
32
h
32
fn
8
o
7
[m+h]+计算值:659.23725;实测:659.23402。
[0135]
实施例10
[0136]
与实施例9的操作步骤相同,只是将靛红换成5-氟靛红,得到1-环丙基-6-氟-7-(4-(2-(4-((5-氟-3-羟亚胺基靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率:56%,其结构式为:
[0137][0138]
1hnmr(400mhz,cdcl3)δ1.02-1.47(7h,m,环丙基-4h和-ch
3
),3.28-3.57(5h,m,哌嗪基-5h),3.73-3.75(4h,m,哌嗪基-1h和-och
3
),4.02-4.04(1h,m,环丙基-1h),4.14-4.16(1h,m,哌嗪基-1h),5.05(2h,d,j=12.0hz,-ch
2-连接基),5.21-5.35(2h,m,-ch
2-连接基),6.97-7.14(3h,m,ar-h),7.79-7.95(2h,m,ar-h),8.85(1h,s,ar-h),14.68(1h,brs,cooh)。
[0139]
hrms-esi:m/z c
32
h
31
f
2
n
8
o
7
[m+h]+计算值:677.22783;实测值:677.22512。
[0140]
实施例11
[0141]
与实施例9的操作步骤相同,只是将靛红换成5-甲基靛红,得到1-环丙基-6-氟-7-(4-(2-(4-((3-羟亚胺基-5-甲基靛红-1-基)甲基)-1h-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率:69%,其结构式为:
[0142][0143]
1hnmr(400mhz,dmso-d6)δ1.03-1.41(7h,m,环丙基-4h和ch
3
),2.26(3h,s,-ch3),3.17-3.55(6h,m,哌嗪基-6h),3.73(3h,s,-och
3
),4.16-4.18(2h,m,哌嗪基-1h和环丙基-1h),5.00(2h,s,-ch
2-连接基),5.38-5.67(2h,m,-ch
2-连接基),7.04(1h,d,j=8.0hz,ar-h),7.24(1h,d,j=4.0hz,ar-h),7.80(1h,d,j=12.0hz,ar-h),8.00(1h,s,ar-h),8.73(1h,s,ar-h),14.92(1h,brs,cooh)。
[0144]
hrms-esi:m/z c
33
h
34
fn
8
o
7
[m+h]+计算值:673.25290;实测值:673.25076。
[0145]
实施例12
[0146]
1、n-炔丙基靛红的合成:
[0147]
将3g(0.0205mol)靛红用50mln,n-二甲基甲酰胺(dmf)溶解,再加入2.44g 3-溴丙炔(0.0205mol)于250ml三颈烧瓶中反应,然后加入8.50g碳酸钾(0.0615mol),在常温下搅拌反应,tlc跟踪反应进程,10小时后反应完毕,向三颈烧瓶中加300ml水稀释,150ml乙酸乙酯萃取,用饱和食盐水洗涤,合并有机相,蒸干,得粗品,经硅胶柱层析纯化得2.44g n-炔丙基靛红(0.0114mol);
[0148]
此步骤的反应方程式为:
[0149][0150]
2、n-丙炔基-3-乙氧亚胺基靛红的合成:
[0151]
将1.0mmol n-炔丙基靛红溶在10ml四氢呋喃和水的混合溶液中,加入1.2mmol乙氧胺盐酸盐和3.0mmol碳酸氢钠固体,在室温下搅拌12h,然后加20ml水进行稀释,接着用30ml乙酸乙酯进行萃取,再用饱和食盐水洗涤,合并有机相,蒸干,得粗品,经硅胶柱层析纯化,得n-炔丙基-3-乙氧亚胺基靛红;
[0152]
此步骤的反应方程式为:
[0153][0154]
3、2-叠氮乙酸琥珀酰亚胺活性酯的合成:
[0155]
将100mmol叠氮乙酸溶解在200ml四氢呋喃中,加入120.0mmol二环己基碳二亚胺和105.0mmol n-羟基琥珀酰亚胺,在室温下搅拌12h,过滤,滤液即2-叠氮乙酸琥珀酰亚胺
活性酯的四氢呋喃溶液,无需纯化,直接使用;
[0156]
此步骤的反应方程式为:
[0157][0158]
4、c-7位连有2-叠氮乙酰基的加替沙星衍生物的合成:
[0159]
向100ml dmf中加入20mmol加替沙星、48ml上述2-叠氮乙酸琥珀酰亚胺活性酯的四氢呋喃溶液和100ml dipea,于室温下搅拌24小时。除去溶剂后,残余物用硅胶柱提纯(洗脱剂dcm:meoh=10:1体积比)得c-7位连有2-叠氮乙酰基的加替沙星衍生物;
[0160]
此步骤的反应方程式为:
[0161][0162]
5、乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体的合成:
[0163]
向50ml dmf中加入10mmol 2-叠氮乙酰基的加替沙星衍生物、12mmoln-丙炔基-3-乙烯基靛红-2-酮和1mmol cu(oac)
2
,在50℃下搅拌2h,然后在减压下浓缩,过反相柱,以甲酸为洗脱剂,得到1-环丙基-7-(4-(2-(4-((3-(乙氧基亚胺基靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-6-氟-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率51%;
[0164]
此步骤的反应方程式为:
[0165][0166]
1hnmr(400mhz,dmso-d6)δ1.20-1.41(10h,m,noch
2
ch
3
,环丙基-4h和-ch
3
),3.34-3.42(4h,m,哌嗪基-4h),3.70-3.82(6h,m,-och
3
和哌嗪基-3h),4.45-4.58(3h,m,noch
2
ch
3
和环丙基-1h),5.02(2h,s,-ch
2-连接基),5.52(2h,s,-ch
2-连接基),7.10(1h,t,j=8.0hz,ar-h),7.19(1h,d,j=4.0hz,ar-h),7.46(1h,d,j=8.0hz,ar-h),7.90-7.95(2h,m,ar-h),8.03(1h,s,ar-h),8.68(1h,s,ar-h)。
[0167]
hrms-esi:m/z c
34
h
36
fn
8
o
7
[m+h]+计算值:687.26855;实测值:687.26346。
[0168]
实施例13
[0169]
与实施例12的操作步骤相同,只是将靛红换成5-氟靛红,得到1-环丙基-7-(4-(2-(4-((3-乙氧基亚胺基-5-氟靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率65%,其结构式为:
[0170][0171]
1hnmr(400mhz,cdcl
3
)δ1.00-1.47(10h,m,noch
2
ch
3
,环丙基-4h和-ch
3
),3.27-3.52(5h,m,哌嗪基-5h),3.73-3.75(4h,m,哌嗪基-1h和-och
3
),4.03-4.05(1h,m,环丙基-1h),4.45-4.56(3h,m,noch
2
ch
3
和哌嗪基-1h),5.24(2h,d,j=12.0hz,-ch
2-连接基),5.30-5.36(2h,m,-ch
2-连接基),6.99-7.14(2h,m,ar-h),7.19-7.88(3h,m,ar-h),8.85(1h,s,ar-h),14.69(1h,brs,cooh)。
[0172]
hrms-esi:m/z c
34
h
35
f
2
n
8
o
7
[m+h]+计算值:705.25913;实测:705.25686。
[0173]
实施例14
[0174]
与实施例1的操作步骤相同,只是将靛红换成5-甲基靛红,得到1-环丙基-7-(4-(2-(4-((3-乙氧基亚胺基-5-甲基靛红-1-基)甲基)-1氢-1,2,3-三唑-1-基)乙酰基)-3-甲基哌嗪-1-基)-6-氟-8-甲氧基-4-氧代-1,4-二氢喹啉-3-羧酸,黄色固体,收率69%,其结构式为:
[0175][0176]
1hnmr(400mhz,cdcl
3
)δ1.28-1.50(10h,m,noch
2
ch
3
,环丙基-4h和-ch
3
),2.27(3h,s,-ch
3
),3.28-3.55(5h,m,哌嗪基-5h),3.73-3.75(4h,m,哌嗪基-1h和-och
3
),4.01-4.03(1h,m,环丙基-1h),4.50-4.60(3h,m,noch
2
ch
3
和哌嗪基-1h),5.08(2h,s,-ch
2-连接子),5.17-5.32(2h,m,-ch
2-连接子),7.06(1h,d,j=8.0hz,ar-h),7.20(1h,d,j=8.0hz,ar-h),7.79(2h,s,ar-h),7.94(1h,d,j=12.0hz,ar-h),8.86(1h,s,ar-h)。
[0177]
hrms-esi:m/z c
35
h
38
fn
8
o
7
[m+h]+计算值:701.28420;实测值:701.28039。
[0178]
实施例1-14的乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体、c-7位连有2-叠氮乙酰基的加替沙星衍生物、环丙沙星和万古霉素对抗革兰阳性菌和阴性菌的活性如下表1和表2所示:
[0179]
表1乙酰基连接的环丙沙星-1,2,3-三氮唑-靛红杂合体的抗革兰阳性菌活性
[0180][0181][0182]
表1中,s.a.金黄色葡萄球菌;mssa对甲氧西林敏感的金黄色葡萄球菌;mrsa耐甲氧西林金黄色葡萄球菌;s.e.表皮葡萄球菌;msse甲氧西林敏感的表皮葡萄球菌,mrse,耐甲氧西林表皮葡萄球菌;s.p,肺炎链球菌;e.fa,粪肠球菌;e.fm,粪肠球菌。
[0183]
表2乙酰基连接的环丙沙星-1,2,3-三氮唑-靛红杂合体的抗革兰阴性菌活性
[0184][0185]
表2中,e.co.1大肠杆菌esbls(-);e.co.2大肠杆菌esbls(+);k.p.1肺炎克雷伯菌esbls(-);k.p.2肺炎克雷伯菌肺炎esbls(-);p.a.铜绿假单胞菌;a.c.阴沟肠杆菌;e.c.大肠杆菌;e.a.,产气肠杆菌;s.m.,嗜麦芽芽胞杆菌;s.m.嗜麦芽寡养单孢菌;c.f.枸橼酸盐杆菌。
[0186]
由表1和表2可知,本发明制备的乙酰基连接的加替沙星-1,2,3-三氮唑-靛红杂合体对多种革兰阳性菌和阴性菌具有良好的活性,是潜在的抗菌药物候选物。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips