
 商标分类
商标分类  商标转让
商标转让 
蒽类衍生物、其制备和应用的制作方法
 2021-02-02 16:02:42|
2021-02-02 16:02:42| 409|
409| 起点商标网
起点商标网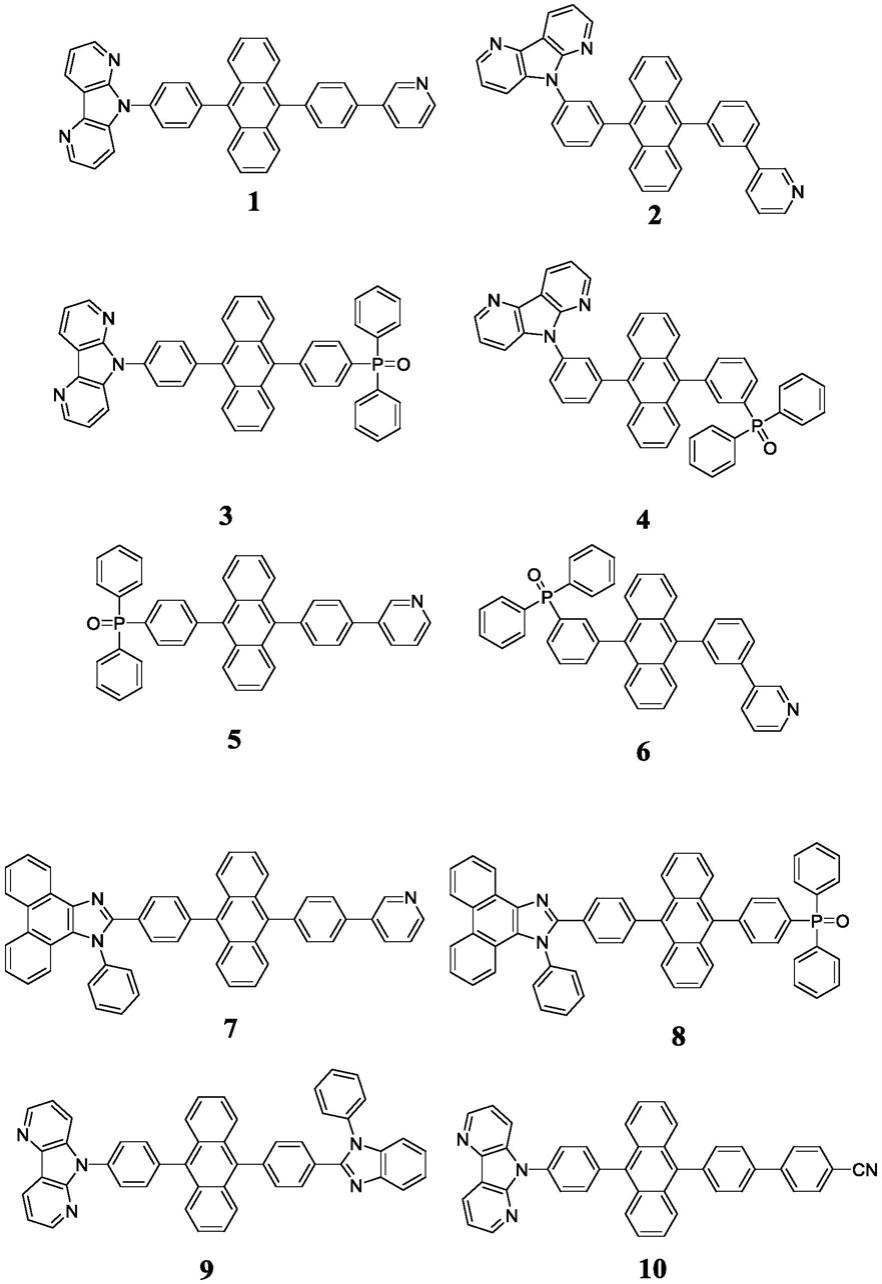
[0001]
本发明属于光电材料应用科技领域,更具体地,涉及一种蒽类衍生物、其制备和应用。
背景技术:
[0002]
自1987年c.w.tang与s.a.vanslyke报道了低驱动电压的三明治结构有机电致发光二极管(oled)(c.w.tang,s.a.vanslyke.organic electroluminescent diodes.appl phys lett,1987,51(12):913-915),关于oled的研究正式步入高速发展的轨道,历经三十多年的发展,oled技术已经成为材料科学和信息技术领域的研究热点。在oled产业化方面,更是展现出了世界范围的“现代高新平板显示技术”国际化竞争态势。为了适应兼顾低成本和高性能的发展要求,需要对oled技术进行更深层次的研究。
[0003]
目前oled还存在瓶颈,蓝光材料一直是其薄弱环节。含有重金属原子的磷光材料尽管可以获得较高的器件效率,但合成成本高,材料稳定性差,导致器件在高亮度下效率滚降严重,器件寿命不足。蓝光荧光材料则具有合成提纯简单,可修饰性强等有点,且荧光器件稳定性更高。但是传统荧光材料也存在一些缺点,比如效率较低,基于掺杂工艺制备器件时为缓解浓度猝灭需要严格控制极低的掺杂浓度,导致工艺难度大幅提高且可重复性变差;深蓝光材料效率不高;材料带隙宽,合适的主体材料较少,能够实现高效能量转移的主-客体系少。(m.zhu,c.yang.blue fluorescent emitters:design tactics and applications in organic light-emitting diodes.chem soc rev 2013,42(12):4963-4976;y.t.chang,j.k.chang,y.t.lee,et al.high-efficiency small-molecule-based organic light emitting devices with solution processes and oxadiazole-based electron transport materials.acs appl mater interfaces 2013,5(21):10614-10622)。具有刚性结构、化学修饰性强、易于提纯、成膜性好等特性的蒽基团,可用于合成蓝光发光材料。杨楚罗教授和马东阁教授合作报道过含蒽基团的深蓝光材料,器件最大亮度3400cd/m
2
,最大电流效率1.80cd/a,cie色度坐标(0.149,0.098)(h.huang,q.fu,s.zhuang,et al.novel deep blue oled emitters with 1,3,5-tri(anthracen-10-yl)benzene-centered starburst oligofluorenes.j phys chem c,2011,115(11):4872-4878)。但是以目前的技术水平来讲,高效蒽类蓝光材料仍然短缺,器件效率不高,光色还有向深蓝区优化的空间。此外,在制备工艺方面如果可以应用到非掺杂发光层结构中,将大幅降低生产成本(h.liu,j.zeng,j.guo,et al.high-performance non-doped oleds with nearly 100%exciton use and negligible efficiency roll-off.angew chem int ed.2018,57(30):9290-9434)。
技术实现要素:
[0004]
针对现有技术的以上缺陷或改进需求,本发明提供了一种蒽类衍生物、其制备和应用,其通过引入弱供电子基团和弱吸电子基团对蒽进行全面修饰获得蒽基衍生物,通过
与不同基团键连实现材料的光电特性,分子轨道能级可调,该类材料可以作为荧光材料、发光主体材料、电子传输材料,应用在有机电致发光领域中,由此解决现有的蒽类衍生物蓝光材料存在的高效率深蓝光色(色度坐标cie y值小于0.1)荧光材料及相应器件短缺的技术问题。
[0005]
为实现上述目的,按照本发明的一个方面,提供了一种1、一种蒽类衍生物,其特征在于,具有如式(一)所示的结构通式:
[0006][0007]
其以蒽为核心,通过取代基团r
1
、r
2
、r
3
和r
4
对蒽进行修饰得到,其中,所述r
1
、r
2
、r
3
和r
4
中至少有一个吸电子基团,且至少有一个供电子基团;
[0008]
通过取代基团r
1
、r
2
、r
3
和r
4
对蒽核心进行修饰,对蒽核心不同活性位点采用不同类型的供吸电子基团以及不同数量的供吸电子基团进行修饰,调控蒽类衍生物电子不同程度的离域,改善蒽类衍生物的能级值、荧光量子产率以及光色,提高其作为发光材料的发光性能。
[0009]
优选地,所述吸电子基团为二苯基磷氧、吡啶、氰基苯、2-苯基菲并咪唑或2-苯基苯并咪唑,所述供电子基团为1,5-氮杂咔唑、2-苯基菲并咪唑或2-苯基苯并咪唑。
[0010]
按照本发明的另一个方面,提供了一种所述的蒽类衍生物的应用,用作有机电致发光器件的有机功能层材料。
[0011]
优选地,用作所述有机功能层中的荧光发光材料、发光主体材料或电子传输材料。
[0012]
按照本发明的另一个方面,提供了一种有机电致发光器件,其有机功能层中包括所述的蒽类衍生物。
[0013]
优选地,所述的有机电致发光器件,其至少有一个电子传输功能层的材料中包含所述的蒽类衍生物。
[0014]
优选地,所述的有机电致发光器件,其电致发光层的材料中包含所述的蒽类衍生物。
[0015]
优选地,所述的有机电致发光器件,包括对电极和设置在该对电极之间的有机发光介质,所述有机发光介质中包含所述的蒽类衍生物。
[0016]
总体而言,通过本发明所构思的以上技术方案与现有技术相比,能够取得下列有益效果:
[0017]
(1)本发明涉及一类全面修饰的蒽类衍生物,选取兼具作为蓝光生色团和高载流子传输性能的蒽作为核心,有利于电子空穴载流子传输平衡和复合,通过在蒽的不同位点键联不同的供电子和吸电子基团,调整外围修饰基团的数目与类型,形成具有多功能的蒽
类衍生物,其可作为荧光发光材料、主体材料和电子传输材料,并可用于制备非掺杂发光层结构的器件中,能够广泛应用于有机发光二级管中,并取得良好的效果,是一种具有很大商业价值的新型材料。
[0018]
(2)本发明通过常用的suzuki反应,ullmann反应等基本反应可以高产率的制备同时用作荧光,发光主体及电子传输材料的多功能蒽类衍生物材料。
[0019]
(3)本发明提供的蒽类衍生物有机材料,将单一材料发挥其多功能特性,制备具有简单同质结构的器件,着力降低生产成本,应用于有机电致发光领域;
[0020]
(4)以本发明提供的蒽类衍生物有机材料为发光体和发光主体,制备的蓝光oled器件性能良好。搭配现阶段商用的基色发光材料制备的白光器件获得了较低的开启电压,较高的效率等优异性能。
[0021]
(5)本发明提供的部分蒽类衍生物有机材料制作有机电致发光器件时,器件电流效率高达5cd/a以上,色度坐标达到(0.14
±
0.01,0.07
±
0.01),发光光色位于深蓝区。此外,在制备工艺方面可以应用到非掺杂发光层结构中,规避了传统的荧光器件采用主-客共掺杂体系中超低掺杂浓度不易控制、工艺重复性差的缺点,同时减少了材料种类的使用,降低了器件制备成本。
附图说明
[0022]
图1是本发明所提供的电致发光器件的结构图。
[0023]
图2所示为器件效率滚降测试结果。
[0024]
图3所示为基于部分化合物所制备的器件光色分布示意图。
[0025]
图4所示为基于部分化合物所制备的器件效率水平示意图。
具体实施方式
[0026]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。此外,下面所描述的本发明各个实施方式中所涉及到的技术特征只要彼此之间未构成冲突就可以相互组合。
[0027]
本发明提供的一种蒽类衍生物,具有如式(一)所示的结构通式:
[0028][0029]
其以蒽为核心,通过取代基团r
1
、r
2
、r
3
和r
4
对蒽进行修饰得到,其中,所述r
1
、r
2
、r
3
和r
4
中至少有一个吸电子基团,且至少有一个供电子基团。
[0030]
本发明通过采用上述取代基团r
1
、r
2
、r
3
和r
4
对蒽进行全面修饰。通过对蒽核心不同活性位点采用不同类型的供吸电子基团以及不同数量的供吸电子基团进行修饰和改进,调控修饰后蒽类衍生物电子不同程度的离域,对蒽类衍生物整体光物理和电化学性能有一定改善,调控的关键参数包括能级值、荧光量子产率、光色等。最终实现了高效率的深蓝光发光材料及器件。
[0031]
本发明所述吸电子基团包括但不限于二苯基磷氧、吡啶、氰基苯、2-苯基菲并咪唑或2-苯基苯并咪唑,所述供电子基团包括但不限于1,5-氮杂咔唑、2-苯基菲并咪唑或2-苯基苯并咪唑。
[0032]
本发明通过引入弱供电子基团和弱吸电子基团对蒽进行全面修饰获得蒽基衍生物,通过与不同基团键连实现材料的光电特性,分子轨道能级可调。本发明所述的弱供电子基团是指具有比咔唑、三苯氨这类常规供电子基团表现出更弱的供电子能力的基团;本发明所述的弱吸电子基团是指比三嗪、蒽醌这类常规的吸电子基团表现出更弱的吸电子能力的基团。减弱供吸电子能力有助于材料实现更蓝的光色。
[0033]
本发明所述的蒽类衍生物包括但不限于以下化合物1~40:,其中化合物1-10核心蒽两个活性位点修饰;11-25核心蒽三个活性位点修饰;26-47核心蒽四个活性位点修饰。
[0034]
[0035]
[0036]
[0037]
[0038]
[0039]
[0040][0041]
本发明所述的蒽类衍生物的制备,可按照常规的有机合成路线进行制备,比如:
[0042]
对于化合物1~10,可通过先对蒽核心两个活性位点进行吸电子、供电子基团取代,采用乌尔曼反应及铃木反应等反应制备得到。
[0043]
对于化合物11~25,可通过先对蒽核心三个活性位点进行吸电子、供电子基团取代,采用乌尔曼反应及铃木反应等反应制备得到。
[0044]
对于化合物26~47,可通过先对蒽核心四个活性位点进行吸电子、供电子基团取代,采用乌尔曼反应及铃木反应等反应制备得到。
[0045]
本发明实施例中列举了以上三类化合物中典型的分子的制备流程,其他化合物分子的制备可根据这些方法或常规的有机合成方法进行设计和调整得到。
[0046]
本发明所提供的蒽类衍生物化合物具有高的荧光量子效率、高的电子载流子迁移率和优秀的材料热稳定性。根据飞行时间法测试材料迁移率可达到2.5
×
10-3
cm
2
/vs至6.0
×
10-3
cm
2
/vs。化合物的热分解温度高,通过tag测定可达到450℃以上,玻璃化转变温度(t
g
)较高,可达到120℃以上。化合物自身蓝光荧光发光效率较高,可应用于蓝光oled器件发光层结构中。同时化合物具备合适的三线态能级,作为主体材料,将黄光、红光和绿光发光染料掺杂之其中后,能够确保激子在发光层中高效复合发光,可应用于白光oled器件的制备。电致发光器件发光效率高,效率滚降小。可广泛应用于电致发光领域。
[0047]
本发明提供的蒽类衍生物,可用作有机电致发光器件的有机功能层材料。在oled器件中可用作其有机功能层中的荧光发光材料、发光主体材料或电子传输材料。本发明的蒽类衍生物有机材料具有适当的三重态、具有良好的载流子迁移特性,在oled器件中可用作发光主体材料或电子传输材料。
[0048]
本发明还提供了一种有机电致发光器件,其有机功能层中包括所述的蒽类衍生物。
[0049]
一些实施例中,有机电致发光器件中至少有一个电子传输功能层的材料中包含本发明所述的蒽类衍生物。
[0050]
本发明的有机材料具有较高的电致发光效率,在有机电致发光显示器中用作电致发光层。一些实施例中,有机电致发光器件的电致发光层的材料中包含所述的蒽类衍生物。
[0051]
一些实施例中,有机电致发光器件包括对电极和设置在该对电极之间的有机发光介质,该有机发光介质中包含本发明所述的蒽类衍生物
[0052]
本发明涉及一类全面修饰的蒽类衍生物及其制备方法、应用和电致发光器件。选取兼具作为蓝光生色团和高载流子传输性能的蒽作为核心,有利于电子空穴载流子传输平衡和复合,通过在蒽的不同位点键联不同的1,5-氮杂咔唑,2-苯基菲并咪唑和吸电子能力的二苯基磷氧,吡啶等,调整外围修饰基团的数目与类型,形成具有多功能的蒽类衍生物,可作为荧光发光材料、主体材料和电子传输材料,并可用于制备非掺杂发光层结构的器件中,能够广泛应用于有机发光二级管中,并取得良好的效果,是一种具有很大商业价值的新型材料。
[0053]
以下为实施例:
[0054]
实施例1
[0055]
化合物1的制备
[0056][0057]
在250ml的单口烧瓶中加入化合物-(5.00g,8.59mmol),3-吡啶硼酸(1.06g,8.59mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.09g,0.08mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物-4.1g,产率90%。ms(apci)m/z calcd for c
31
h
20
in:533.06,found[m+h]+:534.08。
[0058]
在100ml的单口瓶中加入化合物-(2.0g,3.75mmol),-(0.63g,3.75mmol),碘化亚铜(0.06g,0.3mmol),k
2
co
3
(1.38g,10mmol)和18-冠-6(8mg,0.3mmol)溶解到5ml dmpu溶液中。在n
2
的保护下条件下,升温至180℃,反应48h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇20:1过柱子,得到产物(1)1.6g,产率:74%。ms(apci)m/z calcd for c
41
h
26
n
4
:574.22,found[m+h]+:575.24。
[0059]
实施例2
[0060]
化合物3的制备:
[0061][0062]
在100ml的单口瓶中加入-(2.0g,11.8mmol),对溴碘苯(3.33g,11.8mmol),碘化亚铜(0.05g,0.25mmol),k
2
co
3
(3.45g,25mmol)和18-冠-6(6.6mg,0.25mmol)溶解到5ml dmpu溶液中。在n
2
的保护下条件下,升温至180℃,反应48h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇15:1过柱子,得到产物-2.5g,产率:66%。
[0063]
在250ml的单口烧瓶中加入化合物-(1.5g,4.6mmol),9-蒽硼酸(1.1g,5.1mmol),2m的碳酸钾水溶液25ml溶解于25ml乙醇,50ml甲苯的溶剂中。在n2的保护的条件下,加入pd(pph3)4(0.1g,0.1mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物。紧接着将得到的产物(2.1g,5.0mmol),nbs(0.9g,5.5mmol)溶于三氯甲烷(50ml)中。在n
2
的保护下条件下,升温至70℃,反应6h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇20:1过柱子,得到产物
ⅴ
2.1g,产率:84%。ms(apci)m/z calcd for c
30
h
18
brn
3
:499.07,found[m+h]+:500.08.
[0064]
在250ml的单口烧瓶中加入化合物
ⅴ
(2.00g,4.00mmol),化合物
ⅵ
(1.62g,4.00mmol),2m的碳酸钾水溶液20ml溶解于20ml乙醇,40ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph3)4(0.09g,0.08mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物(3)2.0g,产率72%。ms(apci)m/z calcd for c
48
h
32
n
3
op:697.23,found[m+h]+:698.24。
[0065]
实施例3
[0066]
化合物5的制备
[0067][0068]
在100ml的单口瓶中加入化合物-(2.0g,3.75mmol),
ⅶ
(1.9g,9.38mmol),zn(0.7g,11.25mmol),nicl2(h2o)6(0.27g,1.13mmol)和2,2
’-
联吡啶(0.35g,2.26mmol)溶解到5ml dmac溶液中。在n
2
的保护下条件下,升温至180℃,反应48h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇20:1过柱子,得到产物(5)1.7g,产率:75%。ms(apci)m/z calcd for c
43
h
30
nop:607.21,found[m+h]+:608.23。
[0069]
实施例4
[0070]
化合物7的制备
[0071][0072]
在250ml的单口烧瓶中加入化合物-(5.00g,9.38mmol),
ⅷ
(3.17g,9.38mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.05g,0.05mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物(7)5.2g,产率80%。ms(apci)m/z calcd for c
52
h
33
n
3
:699.27,found[m+h]+:700.29。
[0073]
实施例5
[0074]
化合物8的制备
[0075][0076]
在250ml的单口烧瓶中加入化合物-(4.00g,6.87mmol),
ⅷ
(2.32g,6.87mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.04g,0.03mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物
ⅸ
4.6g,产率90%。ms(apci)m/z calcd for c
47
h
29
in
2
:748.14,found[m+h]+:749.18。
[0077]
在100ml的单口瓶中加入化合物
ⅸ
(2.00g,2.67mmol),
ⅶ
(1.35g,6.68mmol),zn(0.51g,8.01mmol),nicl2(h2o)6(0.19g,0.80mmol)和2,2
’-
联吡啶(0.25g,1.60mmol)溶解到10ml dmac溶液中。在n
2
的保护下条件下,升温至180℃,反应48h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇20:1过柱子,得到产物(5)1.6g,产率:73%。ms(apci)m/z calcd for c
59
h
39
n
2
op:822.28,found[m+h]+:823.24。
[0078]
实施例6
[0079]
化合物10的制备
for c
36
h
21
brin
3
:701.00,found[m+h]+:701.03。
[0086]
在250ml的单口烧瓶中加入化合物
ⅻ
(4.00g,5.71mmol),3-吡啶硼酸(1.40g,11.4mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.03g,0.03mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物(11)3.1g,产率83%。ms(apci)m/z calcd for c
46
h
29
n
5
:651.24,found[m+h]+:652.25。
[0087]
实施例8
[0088]
化合物19的制备
[0089][0090]
在250ml的单口烧瓶中加入化合物
ⅺ
(5.00g,7.58mmol),
ⅷ
(2.56g,7.58mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.04g,0.04mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物
ⅹⅲ
5.6g,产率90%。ms(apci)m/z calcd for c
47
h
28
brin
2
:826.05,found[m+h]+:827.08。
[0091]
在250ml的单口烧瓶中加入化合物
ⅹⅲ
(5.00g,6.05mmol),3-吡啶硼酸(1.49g,12.1mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.03g,0.03mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物(19)4.1g,产率87%。ms(apci)m/z calcd for c
57
h
36
n
4
:766.29,found[m+h]+:767.31。
[0092]
实施例9
[0093]
化合物21的制备
calcd for c
48
h
33
n
2
op:684.23,found[m+h]+:685.26。
[0101]
实施例11
[0102]
化合物26的制备
[0103][0104]
在100ml的单口瓶中加入化合物
ⅹⅴ
(3.0g,4.05mmol),-(0.68g,4.05mmol),碘化亚铜(0.08g,0.41mmol),k
2
co
3
(1.12g,8.10mmol)和18-冠-6(0.11g,0.41mmol)溶解到8ml dmpu溶液中。在n
2
的保护下条件下,升温至180℃,反应48h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇20:1过柱子,得到产物
ⅹⅵ
2.4g,产率:76%。ms(apci)m/z calcd for c
36
h
20
br
2
in
3
:780.90,found[m+h]+:781.91。
[0105]
在250ml的单口烧瓶中加入化合物
ⅹⅵ
(3.00g,3.84mmol),3-吡啶硼酸(1.42g,11.5mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.02g,0.02mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物(26)2.5g,产率89%。ms(apci)m/z calcd for c
51
h
32
n
6
:728.27,found[m+h]+:729.28。
[0106]
实施例12
[0107]
化合物42的制备
[0108][0109]
在250ml的单口烧瓶中加入化合物
ⅹⅴ
(5.00g,6.76mmol),
ⅷ
(2.29g,6.76mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.03g,0.03mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物
ⅹⅶ
4.9g,产率80%。ms(apci)m/z calcd for c
47
h
27
br
2
in
2
:905.96,found[m+h]+:906.98。
[0110]
在250ml的单口烧瓶中加入化合物
ⅹⅶ
(4.00g,4.42mmol),3-吡啶硼酸(1.63g,13.26mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.02g,0.02mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物(42)3.2g,产率85%。ms(apci)m/z calcd for c
62
h
39
n
5
:853.32,found[m+h]+:854.35。
[0111]
实施例13
[0112]
化合物44的制备
[0113][0114]
在250ml的单口烧瓶中加入化合物
ⅹⅴ
(5.00g,6.76mmol),3-吡啶硼酸(1.66g,13.52mmol),2m的碳酸钾水溶液30ml溶解于30ml乙醇,60ml甲苯的溶剂中。在n
2
的保护的条件下,加入pd(pph
3
)
4
(0.04g,0.03mmol)。缓慢升温至110℃,混合物在回流状态下反应24h。冷却后分液,旋蒸有机层,柱层析,得到产物
ⅹⅷ
3.8g,产率88%。ms(apci)m/z calcd for c
36
h
22
br
2
n
2
:642.01,found[m+h]+:643.02。
[0115]
在100ml的单口瓶中加入化合物
ⅹⅷ
(3.0g,4.67mmol),-(1.58g,9.35mmol),碘化亚铜(0.09g,0.5mmol),k
2
co
3
(1.29g,9.35mmol)和18-冠-6(0.13g,0.5mmol)溶解到8ml dmpu溶液中。在n
2
的保护下条件下,升温至180℃,反应48h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇20:1过柱子,得到产物(44)2.7g,产率:74%。ms(apci)m/z calcd for c
56
h
38
n
8
:818.29,found[m+h]+:819.30。
[0116]
实施例14
[0117]
化合物46的制备
[0118]
[0119]
在100ml的单口瓶中加入化合物
ⅹⅷ
(2.0g,3.11mmol),
ⅶ
(3.15g,15.58mmol),锌粉(0.61g,9.33mmol),nicl
2
(h
2
o)6(0.2g,0.9mmol)和2,2
’-
联吡啶(0.28g,1.8mmol)溶解到8ml dmac溶液中。在n2的保护下条件下,升温至170℃,反应48h。待反应终止后,冷却至室温,萃取,旋干,二氯甲烷:甲醇20:1过柱子,得到产物(23)2.1g,产率:76%。ms(apci)m/z calcd for c
60
h
42
n
2
o
2
p
2
:884.27,found[m+h]+:885.29。
[0120]
实施例15:
[0121]
化合物1作为发光层制备器件。图1为器件结构示意图,采用玻璃基底,复合氧化金属薄膜氧化铟锡(ito)作为器件阳极,该复合氧化金属薄膜可以采用磁控溅射的方法制备在基板之上(当然,也可以直接采用市售ito玻璃商品)。ito上采用热蒸镀的方法制备氧化钼(moo
3
)薄膜作为空穴注入层,tapc作为空穴传输层,mcp作为激子阻挡层拥有较高的三线态能级(t
1
)可以有效的将激子限制在发光层中,提高辐射复合几率,提高器件效率。发光层采用非掺杂结构,发光材料为本发明所提供的一系列蒽类修饰衍生物。在发光层与电子注入层liq之间生长一层tpbi作为电子传输层,金属铝(al)作为阴极。除本发明提供的首创材料外,其余功能层材料均为商业化材料,可通过市场采购获得。
[0122]
这个实例展示了1作为发光材料而制备的电致发光器件的性能验证。150nm的ito(氧化铟锡)玻璃相继在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(105℃),再将ito玻璃放入等离子反应器中进行5分钟的氧等离子处理,传送到真空室内制备有机膜和金属电极,接着通过真空蒸镀的方法制备一层10nm的空穴注入材料三氧化钼,接着蒸镀80nm厚的空穴传输材料:4,4'-环己基二[n,n-二(4-甲基苯基)苯胺](tapc),然后蒸镀10nm厚的激子阻挡层1,3-二-9-咔唑基苯(mcp),再通过真空蒸镀20nm的发光层1化合物,之后蒸镀1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)30nm作为电子传输层,最后再蒸镀一层1nm的liq和100nm的al金属电极。
[0123]
器件性能评价:将铝作为器件的阴极,将直流电的正极加于ito(氧化铟锡),将负极加于金属层。使用电脑控制的凯瑟琳2400(keithley 2400)数字源表测量电流-电压特性,使用光谱扫描(spectrascan pr655)亮度计来评价发光性能。器件的表现为启亮电压2.8v,最大电流效率为6.1cd/a,4.5lm/w,亮度在100cd/m
2
时的cie色坐标为(0.14,0.06)。亮度达到1000cd/m
2
时的器件效率几乎没有滚降。随电压增加,光谱稳定性良好,如图2所示。
[0124]
实施例16:
[0125]
化合物35作为发光层制备器件。
[0126]
这个实例展示了35作为发光材料而制备的电致发光器件的性能验证。150nm的ito(氧化铟锡)玻璃相继在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(105℃),再将ito玻璃放入等离子反应器中进行5分钟的氧等离子处理,传送到真空室内制备有机膜和金属电极,接着通过真空蒸镀的方法制备一层10nm的空穴注入材料三氧化钼,接着蒸镀80nm厚的空穴传输材料:4,4'-环己基二[n,n-二(4-甲基苯基)苯胺](tapc),然后蒸镀10nm厚的激子阻挡层1,3-二-9-咔唑基苯(mcp),再通过真空蒸镀20nm的发光层35化合物,之后蒸镀1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)30nm作为电子传输层,最后再蒸镀一层1nm的liq和100nm的al金属电极。
[0127]
器件性能评价:将铝作为器件的阴极,将直流电的正极加于ito(氧化铟锡),将负
极加于金属层。使用电脑控制的凯瑟琳2400(keithley 2400)数字源表测量电流-电压特性,使用光谱扫描(spectrascan pr655)亮度计来评价发光性能。器件的表现为启亮电压2.8v,最大电流效率为5.1cd/a,4.0lm/w,亮度在100cd/m
2
时的cie色坐标为(0.14,0.05)。亮度达到1000cd/m
2
时的器件效率几乎没有滚降。随电压增加,光谱稳定性良好。
[0128]
实施例17
[0129]
化合物7作为发光层主体材料制备互补色白光器件。
[0130]
这个实例展示了35作为发光材料而制备的电致发光器件的性能验证。150nm的ito(氧化铟锡)玻璃相继在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(105℃),再将ito玻璃放入等离子反应器中进行5分钟的氧等离子处理,传送到真空室内制备有机膜和金属电极,接着通过真空蒸镀的方法制备一层10nm的空穴注入材料三氧化钼,接着蒸镀80nm厚的空穴传输材料:4,4'-环己基二[n,n-二(4-甲基苯基)苯胺](tapc),然后蒸镀10nm厚的激子阻挡层1,3-二-9-咔唑基苯(mcp),再通过真空蒸镀20nm的发光层,先蒸镀10nm厚的7化合物作蓝光发光单元,再蒸镀10nm化合物7:掺杂黄光染料1.0wt%tbrb的黄光发光单元,之后蒸镀1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯(tpbi)30nm作为电子传输层,最后再蒸镀一层1nm的liq和100nm的al金属电极。
[0131]
器件性能评价:将铝作为器件的阴极,将直流电的正极加于ito(氧化铟锡),将负极加于金属层。使用电脑控制的凯瑟琳2400(keithley 2400)数字源表测量电流-电压特性,使用光谱扫描(spectrascan pr655)亮度计来评价发光性能。器件的表现为启亮电压3.0v,最大电流效率为15.4cd/a,14.1lm/w,亮度在100cd/m
2
时的cie色坐标为(0.38,0.43)。亮度达到1000cd/m
2
时的电流效率滚降为6.9%,功率效率滚降为13.0%。显色指数cri值为58。随电压增加,光谱稳定性良好。
[0132]
实施例18:
[0133]
4作为发光层和电子传输层制备器件。
[0134]
这个实例展示了4兼作为发光材料和电子传输材料而制备的电致发光器件的性能验证。150nm的ito(氧化铟锡)玻璃相继在清洗剂和去离子水中以超声波清洗30分钟。然后真空干燥2小时(105℃),再将ito玻璃放入等离子反应器中进行5分钟的氧等离子处理,传送到真空室内制备有机膜和金属电极,接着通过真空蒸镀的方法制备一层10nm的空穴注入材料三氧化钼,接着蒸镀80nm厚的空穴传输材料:4,4'-环己基二[n,n-二(4-甲基苯基)苯胺](tapc),然后蒸镀5nm厚的激子阻挡层1,3-二-9-咔唑基苯(mcp),再通过真空蒸镀50nm的4化合物兼作为发光层和电子传输层,最后再蒸镀一层1nm的liq和100nm的al金属电极。
[0135]
器件性能评价:将铝作为器件的阴极,将直流电的正极加于ito(氧化铟锡),将负极加于金属层。使用电脑控制的凯瑟琳2400(keithley 2400)数字源表测量电流-电压特性,使用光谱扫描(spectrascan pr655)亮度计来评价发光性能。器件的表现为启亮电压3.2v,最大电流效率为4.5cd/a,3.5lm/w,亮度在100cd/m
2
时的cie色坐标为(0.15,0.06)。亮度达到1000cd/m
2
时的器件效率几乎没有滚降。随电压增加,光谱稳定性良好。
[0136]
化合物1,2,3,5,11,14,30,35,36,40分别制备电致发光器件,厚度单位nm。器件性能见下表1。表1是部分化合物的在有机电致发光器件中表现,从表中数据可以看出该类材料在作为荧光发光材料时,器件效率较高,电流效率可达到5cd/a以上,色度坐标值达到(0.14
±
0.01,0.07
±
0.01),光色位于深蓝光区域,此时对应的器件外量子效率达到4.5%
以上,接近荧光材料外量子效率理论极限(5%),是一类很有应用前景的发光材料。同时可以用于制备白光有机电致发光器件以拓宽其应用领域范围。图3所示为基于部分化合物所制备的器件光色分布示意图。图4所示为基于部分化合物所制备的器件效率水平示意图,横坐标对应各化合物在本发明中的数字编号。
[0137]
表1.部分化合物的器件基本性质
[0138]
[0139][0140]
由上可知以本发明提供的一类全面修饰的蒽类衍生物及所制备的电致发光器件具有开启电压低,电流效率高,且光色达到了深蓝光区域(色度坐标cie y值基本在0.07以下,甚至可以达到0.05)。依据(美国)国家电视标准委员会的ntsc标准(national television system committee)定义的标准蓝光cie(x,y)(0.14,0.08)以及欧洲广播联盟的ebu标准(european broadcasting union)定义的标准蓝光cie(x,y)(0.15,0.06),均可得到结论,本发明提供的新型蒽类衍生物具备深蓝光发光特性。同时材料具备多种功能,可应用在发光层结构和电子传输层结构中。本发明所提供的技术方案在构建蓝光有机发光材料及相关电致发光器件上有重要的应用价值。
[0141]
本领域的技术人员容易理解,以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips