
 商标分类
商标分类  商标转让
商标转让 
一种稳定同位素氘标记的盐酸金刚乙胺及其合成方法与流程
 2021-02-02 16:02:18|
2021-02-02 16:02:18| 372|
372| 起点商标网
起点商标网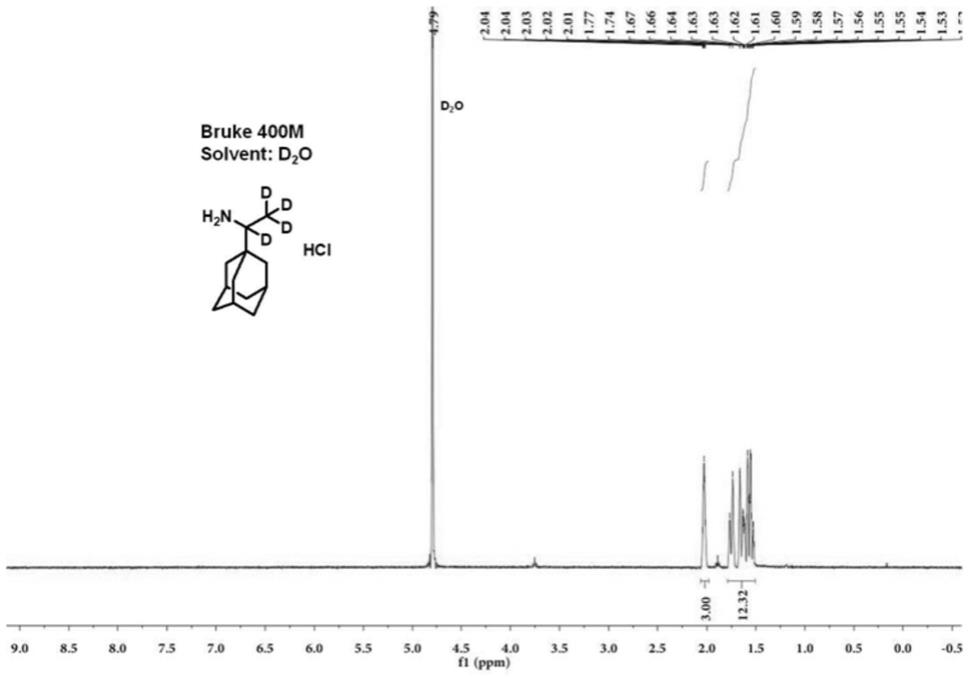
[0001]
本发明属于精细化工合成技术领域,特别是涉及一种稳定同位素氘标记的盐酸金刚乙胺及其合成方法。
背景技术:
[0002]
盐酸金刚乙胺,由美国bristol-myers squibb公司研发,1987年首次在法国上市,1993年美国fda批准用于预防和治疗a型流感病毒感染,在临床上,也用于治疗突然的剧痛和麻疹,其特点是吸收快且完全、毒副作用小。
[0003]
对于盐酸金刚乙胺的定量分析,传统的检测方法主要有高效液相色谱法(hplc)、液相色谱-质谱法(hplc/ms)、薄层色谱法(tlc)、气相色谱法(gc)、气相色谱-质谱联机(gc/ms)、毛细管电泳(ce)等,但是这些方法普遍存在灵敏度低,方法繁琐,或只能进行半定量分析等缺点。
[0004]
稳定同位素稀释质谱法idms(isotope dilution mass spectrometry)是采用与被测物质具有相同分子结构的稳定同位素标记的化合物作为内标物质,用高分辨液相色谱-质谱联用仪(lc-ms)进行检测,通过质谱仪测量相应质量数的离子的比值并与标准的比值比较达到准确定量的目的。采用同位素内标可以有效消除样品在化学和物理的前处理步骤中所引起的回收率差异,从而避免处理样品过程中的损失对检测结果造成的偏差。稳定同位素内标的特性与lc-ms的高灵敏度和处理复杂样品的能力结合,使得色谱-同位素稀释质谱技术被公认为测量微量及痕量有机物的基准定量方法,应用越来越广泛。
[0005]
目前,由于稳定同位素标记原料价格较贵,成本较高、生产工艺较难实现、如何保证生产过程中稳定同位素原子不会脱落、产品纯化等技术难点,稳定同位素标记的盐酸金刚乙胺的合成方法尚未见报道。因此,有必要提供一种稳定同位素标记的盐酸金刚乙胺的合成方法,能得到化学纯度和同位素丰度均非常高的稳定同位素标记的盐酸金刚乙胺,作为稳定同位素内标用于idms方法,定量分析盐酸金刚乙胺。
技术实现要素:
[0006]
本发明所要解决的技术问题是提供一种稳定同位素氘标记的盐酸金刚乙胺及其合成方法,可以作为定量检测盐酸金刚乙胺的标准试剂;且制备过程简单,稳定同位素氘不会脱落,产品容易分离提纯,得到的产品化学纯度和同位素丰度高。
[0007]
本发明为解决上述技术问题而采用的技术方案是提供一种稳定同位素氘标记的盐酸金刚乙胺的合成方法,包括以下步骤:s1:将金刚烷甲酰氯与氘代碘甲烷反应,制得氘代金刚烷甲基酮;s2:将氘代金刚烷甲基酮与盐酸羟胺反应,制得氘代金刚烷甲基酮肟;s3:氘代金刚烷甲基酮肟经氘化铝锂还原、加盐酸成盐,制得稳定同位素氘标记的盐酸金刚乙胺。
[0008]
进一步地,所述步骤s1过程如下:先将氘代碘甲烷和镁在乙醚中,25~30℃温度下反应1~2小时制得氘代甲基碘化镁;然后在惰性气体保护下,加入到金刚烷甲酰氯和氯化
亚铜的乙醚溶液中,25~30℃温度下反应0.5~1小时,2mol/l盐酸溶液淬灭,乙醚萃取,柱层析得到氘代金刚烷甲基酮。
[0009]
进一步地,所述氘代碘甲烷和镁的摩尔比为1:1~1:1.5,在保证氘代碘甲烷完全反应的同时节约镁的用量;所述惰性气体为氮气或氩气;所述氘代碘甲烷和金刚烷甲酰氯的摩尔比为1:0.8~1:1.2;所述氘代碘甲烷和氯化亚铜的摩尔比为1:0.04~1:0.06;所述氘代甲基碘化镁的乙醚溶液加入过程为1~2滴/秒逐滴滴加,且在冰盐浴下进行,温度保持在0~5℃,以防止局部过热导致副反应的发生,从而提高氘代金刚烷甲基酮的产率。
[0010]
进一步地,所述步骤s2过程如下:氘代金刚烷甲基酮、盐酸羟胺和乙酸钠在水和乙醇的混合溶剂中,80~100℃温度下反应2~4小时,冷却后过滤,滤饼水洗至ph7.0,70~80℃干燥2~3小时,得到氘代金刚烷甲基酮肟。
[0011]
进一步地,所述氘代金刚烷甲基酮和盐酸羟胺的摩尔比为1:1~1:1.5;所述氘代金刚烷甲基酮和乙酸钠的摩尔比为1:2~1:2.5。
[0012]
进一步地,所述步骤s3过程如下:氘代金刚烷甲基酮肟溶于四氢呋喃溶剂中,加入氘化铝锂,70~80℃温度下反应4~8小时,恢复至25~30℃后淬灭,25~30℃温度下搅拌2~4小时,过滤,甲醇洗涤,减压去除溶剂四氢呋喃和甲醇,加入浓盐酸成盐,得到稳定同位素氘标记的盐酸金刚乙胺。
[0013]
进一步地,所述氘代金刚烷甲基酮肟和氘化铝锂的摩尔比为1:5~1:10;所述四氢呋喃为干燥无水的溶剂;所述氘化铝锂加入过程在冰盐浴下进行,温度保持在0~5℃,以防止局部过热导致副反应的发生;所述的淬灭过程为依次加入水、15~20wt%氢氧化钠、水。
[0014]
本发明为解决上述技术问题还提供一种根据上述合成方法制备得到的稳定同位素氘标记的盐酸金刚乙胺,其分子结构为:
[0015][0016]
本发明具有以下优点:
[0017]
(1)本发明合成工艺路线短,过程简单,条件温和,稳定同位素氘不会脱落,能得到特定位置稳定同位素氘标记的产品。
[0018]
(2)本发明产品容易分离提纯,产率高,产品化学纯度与稳定同位素丰度均达到99%以上,可充分满足定量检测金刚乙胺的标准试剂的要求。
[0019]
(3)本发明使用价值高、具有良好的经济性。
附图说明
[0020]
图1为实施例1得到的稳定同位素氘标记的盐酸金刚乙胺-d
4
的核磁共振氢谱图。
[0021]
图2为实施例1得到的稳定同位素氘标记的盐酸金刚乙胺-d
4
的高效液相色谱图。
具体实施方式
[0022]
下面结合实施例对本发明作进一步的描述,但不应理解为是对本发明的限制。
[0023]
实施例1
[0024]
稳定同位素氘标记的盐酸金刚乙胺的合成过程如下:
[0025]
s1、氘代碘甲烷(15mmol,2.2g)和镁(18mmol,438mg)在乙醚溶液中,25~30℃温度下反应1.5小时制得氘代甲基碘化镁,在氮气保护,0~5℃温度下,以1滴/秒的速度滴加到金刚烷甲酰氯(13mmol,2.6g)和氯化亚铜(0.72mmol,72mg)的乙醚溶液中,25~30℃温度下反应0.5小时,2m盐酸溶液淬灭,乙醚萃取,柱层析得到氘代金刚烷甲基酮,产率80%;
[0026]
s2、氘代金刚烷甲基酮(5.5mmol,980mg)、盐酸羟胺(8mmol,556mg)和乙酸钠(12mmol,984mg)在水和乙醇的混合溶剂中,85℃温度下反应4小时,冷却后过滤,滤饼水洗至ph7.0,70~80℃干燥2~3小时,得到氘代金刚烷甲基酮肟,产率90%;
[0027]
s3、氘代金刚烷甲基酮肟(5mmol,1g)溶于干燥的四氢呋喃溶剂中,0~5℃温度下,加入氘化铝锂(40mmol,1.68g),80℃温度下反应7小时,恢复至25~30℃后依次加入水、15~20wt%氢氧化钠、水淬灭,25~30℃温度下搅拌3小时,过滤,甲醇洗涤,减压去除溶剂四氢呋喃和甲醇,加入浓盐酸成盐,得到稳定同位素氘标记的盐酸金刚乙胺,产率60%,所得产品的化学纯度以及稳定同位素丰度均达到99%以上。
[0028]
本实施例获取的产品以d
2
o为溶剂,通过bruke-400m核磁共振仪器检测得到图1所示的核磁共振氢谱图,从图1可以看出,化学位移~1.2ppm和~3.0ppm处未见吸收峰,表明结构为盐酸金刚乙胺-d
4
。
[0029]
同时,本实施例获取的产品样品溶于乙腈/水(体积比1:1),以乙腈:0.06wt%三氯乙酸溶液=33:67为流动相,1.0ml/min的流速通过柱温为30℃的液相柱(diamonsil c18 4.6*150mm,5um),通过elsd检测器获得盐酸金刚乙胺-d
4
的高效液相色谱图,如图2所示,从图2可以看出,样品纯度达到99%以上。
[0030]
实施例2
[0031]
稳定同位素氘标记的盐酸金刚乙胺的合成过程如下:
[0032]
s1、氘代碘甲烷(13mmol,1.85g)和镁(15mmol,365mg)在乙醚溶液中,25~30℃温度下反应2小时制得氘代甲基碘化镁,在氩气保护,0~5℃温度下,以2滴/秒的速度滴加到金刚烷甲酰氯(15mmol,2.98g)和氯化亚铜(0.7mmol,70mg)的乙醚溶液中,25~30℃温度下反应1小时,2m盐酸溶液淬灭,乙醚萃取,柱层析得到氘代金刚烷甲基酮,产率85%;
[0033]
s2、氘代金刚烷甲基酮(5mmol,896mg)、盐酸羟胺(7.5mmol,522mg)和乙酸钠(12mmol,984mg)在水和乙醇的混合溶剂中,90℃温度下反应3小时,冷却后过滤,滤饼水洗至ph7.0,70~80℃干燥2~3小时,得到氘代金刚烷甲基酮肟,产率88%;
[0034]
s3、氘代金刚烷甲基酮肟(4.4mmol,880mg)溶于干燥的四氢呋喃溶剂中,0~5℃温度下,加入氘化铝锂(30mmol,1.26g),85℃温度下反应6小时,恢复至25~30℃后依次加入水、15~20wt%氢氧化钠、水淬灭,25~30℃温度下搅拌4小时,过滤,甲醇洗涤,减压去除溶剂四氢呋喃和甲醇,加入浓盐酸成盐,得到稳定同位素氘标记的盐酸金刚乙胺,产率58%,所得产品的化学纯度以及稳定同位素丰度均达到99%以上。
[0035]
虽然本发明已以较佳实施例揭示如上,然其并非用以限定本发明,任何本领域技术人员,在不脱离本发明的精神和范围内,当可作些许的修改和完善,因此本发明的保护范
围当以权利要求书所界定的为准。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips