
 商标分类
商标分类  商标转让
商标转让 
一种以1,6-己二醇和三聚氯氰为原料合成6-氯-1-己醇的方法与流程
 2021-02-02 16:02:38|
2021-02-02 16:02:38| 348|
348| 起点商标网
起点商标网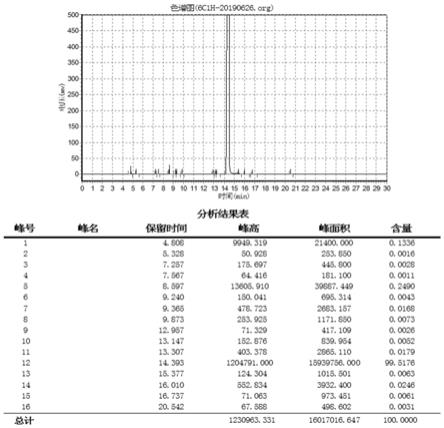
[0001]
本发明涉及有机合成领域,尤其涉及一种以1,6-己二醇和三聚氯氰为原料合成6-氯-1-己醇的方法。
背景技术:
[0002]
在现有技术中,合成6-氯-1-己醇的方法有多种,其中最重要的是利用1,6-己二醇作为原料,使用氯代试剂来取代。主要方法有:(1)选择以六羰基钼作为催化试剂,以四氯化碳作为氯化试剂(khusnutdinov; shchadneva; burangulova; muslimov; dzhemilev russian journal of organic chemistry, 2006 , vol. 42, # 11 p. 1615
ꢀ-ꢀ
1621)。文献报道该方法合成所得的6-氯-1-己醇收率较高,但是其缺点是六羰基钼售价昂贵,供应商少,同时四氯化碳毒害性较大,为人类可疑致癌物,是我国环境优先控制物之一,因此在上述各不利因素下,上述方法并不适合大规模的工业化生产。
[0003]
(2)采用浓盐酸作为氯代试剂(us5237113, 1993, a),该方法的缺点是收率较低,只有82%左右,同时反应过程中杂质较多,生成较多量的1,6-二氯己烷和缩合产物。并且该方法所产生的酸性废水量较大,污染较大,因此该方法同样不适于大规模工业化生产。
技术实现要素:
[0004]
为了解决上述技术问题,本发明提供了一种以1,6-己二醇和三聚氯氰为原料合成6-氯-1-己醇的方法,本发明以1,6-己二醇和三聚氯氰为原料合成6-氯-1-己醇,原料便宜易得,所得产物纯度可达99%以上,产率可达95%以上,反应条件温和,后处理简单,更加适于大规模的工业化应用。
[0005]
本发明的具体技术方案为:一种以1,6-己二醇和三聚氯氰为原料合成6-氯-1-己醇的方法,包括以下步骤:1)将三聚氯氰溶于溶剂a中,搅拌反应,反应完毕后,所得溶液备用。
[0006]
2)配制1,6-己二醇或其溶液。
[0007]
3)将步骤1)所得溶液慢慢滴加到步骤2)所得的1,6-己二醇或其溶液中去,滴加完毕后保温反应,反应完成后将反应液过滤,用溶剂b漂洗滤饼,收集滤液。
[0008]
4)将步骤3)所得滤液精馏,制得成品产品。
[0009]
本发明的反应原理如下:使用三聚氯氰和n,n-二甲基苯胺、n,n-二甲基甲酰胺和二甲基亚砜等溶剂首先生成活化的氯代试剂(具体的活化机理是三聚氯氰首先和n,n-二甲基甲酰胺等溶剂发生亲核取代反应,生成活泼型的n,n-二甲基氯代产物,后者和醇羟基反应,脱去氯化氢,形成不稳定的中间体,后者与氯离子发生亲核取代生成产物),然后再将其滴加到1,6-己二醇溶液中,使反应过程中1,6-己二醇始终处于过量状态,从而能够限制副产物二氯己烷的生成和其他缩合产物的生成。本发明方法收率、纯度高,成本低,更适于实
际工业化推广。
[0010]
从上述机理可知,本发明能够实现高收率、高纯度以及可实现温和反应的原因在于:原料醇不是直接和三聚氯氰反应,而是三聚氯氰先和溶剂反应生成活泼型较高的n,n-二甲基氯代产物,再和原料醇反应,使得反应条件降低。由于生产的中间体体积较大,所以不容易生成二氯的取代产物。传统的氯代是直接由氯离子取进行亲核取代,对于一般的单取代醇是比较好的。
[0011]
此外,需要说明的是,虽然本发明选用了新的氯代试剂,但是若要进一步实现高纯度和高产率,仍需要严格配合工艺参数以及其他试剂的选择。为此,本发明团队针对本发明体系经过大量研究后研发了以下整套具体工艺,能够取得理想的技术效果。
[0012]
作为优选,步骤1)中,所述溶剂a选自n,n-二甲基苯胺、n,n-二甲基甲酰胺和二甲基亚砜。
[0013]
溶剂a需要严格筛选,本发明团队经过大量试验,发现只有在上述溶剂下才可获得最佳的技术效果。
[0014]
作为优选,步骤1)中,所述溶剂a和三聚氯氰的重量比为2~10:1。
[0015]
本发明选用上述范围在于,若比例低于上述比值,物料反应后粘度过大,搅拌困难。若高于上述比值,溶剂浪费。
[0016]
作为优选,步骤1)中,反应温度为10-25℃。
[0017]
若反应温度过低,造成反应速度过慢,若反应温度过高,容易造成杂质生成。
[0018]
作为优选,步骤2)中,所述溶液采用的溶剂为n,n-二甲基苯胺、n,n-二甲基甲酰胺、二甲基亚砜、二氯甲烷或甲苯。
[0019]
本发明将溶剂进一步限定为上述范围,上述溶剂对其溶解度比较高,或者和步骤1)中溶剂相同,使反应体系只有一种溶剂,方便后处理。
[0020]
作为优选,所述溶液采用的溶剂和1,6-己二醇的重量比为1~3:1。
[0021]
1,6-己二醇常温下属于固体,上述溶剂对其溶解度的高低,在上述配比下效果最佳。
[0022]
作为优选,步骤3)中,所述1,6-己二醇和三聚氯氰的摩尔比为0.95-1.05:1。
[0023]
本发明团队在实验中发现,上述摩尔比非常重要,若摩尔比太低,造成三聚氯氰过量,可能产生杂质,后处理麻烦。同时浪费原料,浪费溶剂。若摩尔比太高,造成原料己二醇过量,浪费原料,浪费溶剂。
[0024]
作为优选,步骤3)中,滴加过程的温度控制在-5-25℃,反应时间为1-8h。
[0025]
若温度太低,反应太慢,时间效益不好、若温度太高,会生成杂质的概率提高,使产品精制麻烦。
[0026]
作为优选,步骤3)中,过滤为低真空抽滤或甩滤;所述溶剂和步骤2)中所用溶剂相同;所述溶剂b的用量为三聚氯氰重量的5-15%。
[0027]
作为优选,步骤4)中,所述精馏包括常压精馏和减压高真空精馏,其中所述减压高真空精馏的真空度在3-10mmhg之间。
[0028]
与现有技术对比,本发明的有益效果是:本发明以1,6-己二醇和三聚氯氰为原料合成6-氯-1-己醇,原料便宜易得,所得产物纯度可达99%以上,产率可达95%以上,反应条件温和,后处理简单,更加适于大规模的工业化应用。
附图说明
[0029]
图1为实施例1所得产物的gc结果图;图2为实施例1所得产物的h-nmr图。
具体实施方式
[0030]
下面结合实施例对本发明作进一步的描述。
[0031]
总实施例一种以1,6-己二醇和三聚氯氰为原料合成6-氯-1-己醇的方法,包括以下步骤:1)将三聚氯氰溶于溶剂a(n,n-二甲基苯胺、n,n-二甲基甲酰胺或二甲基亚砜)中,溶剂a和三聚氯氰的重量比为2~10:1,10-25℃下搅拌反应,反应完毕后,所得溶液备用。
[0032]
2)配制1,6-己二醇或其溶液。溶液采用的溶剂为n,n-二甲基苯胺、n,n-二甲基甲酰胺、二甲基亚砜、二氯甲烷或甲苯,溶剂和1,6-己二醇的重量比为1~3:1。
[0033]
3)将步骤1)所得溶液慢慢滴加到步骤2)所得的1,6-己二醇或其溶液中去(期间保持-5-25℃),1,6-己二醇和三聚氯氰的摩尔比为0.95-1.05:1,滴加完毕后保温反应1-8h,反应完成后将反应液过滤(过滤为低真空抽滤或甩滤),用溶剂b(和步骤2)中所用溶剂相同)漂洗滤饼,溶剂b的用量为三聚氯氰重量的5-15%,收集滤液。
[0034]
4)将步骤3)所得滤液精馏,精馏包括常压精馏和减压高真空精馏,其中所述减压高真空精馏的真空度在3-10mmhg之间,最后制得成品产品。
[0035]
实施例11)四口瓶中加入3.7kg dmf,搅拌,温度控制在10-20℃,分批次加入738g三聚氯氰,加入完毕,搅拌6h,检测三聚氯氰已经反应完毕。
[0036]
2)配制473g 1,6-己二醇和500g的dmf的混合溶液,搅拌,将温度控制在-5-0℃,滴加1)中所配反应液,滴加完毕,0℃下搅拌反应2小时,停止制冷,自然升温至25℃,取样中控,当1,6-己二醇完全反应,反应结束。
[0037]
3)将步骤3)所得的混合反应液低真空抽滤,滤饼用100g的dmf漂洗,抽干,合并产品的dmf溶液。
[0038]
4)减压精馏产品,控制真空度在5mmhg,缓慢升温精馏,得产品:520g,纯度99.52%:收率:95.2%。本实施例所得产物的gc图谱以及h-nmr分别如图1-2所示。
[0039]
实施例21)四口瓶中加入3.7kg 二甲基亚砜,搅拌,温度控制在10-20℃,分批次加入738g三聚氯氰,加入完毕,搅拌2h,检测三聚氯氰已经反应完毕。
[0040]
2)配制473g 1,6-己二醇和700g的二甲基亚砜的混合溶液,搅拌,将温度控制在0-10℃,滴加1)中所配反应液,滴加完毕,10℃下搅拌反应2小时,停止制冷,自然升温至25℃,取样中控,当1,6-己二醇完全反应,反应结束。
[0041]
3)将上述所得的混合反应液低真空抽滤,滤饼用100g的二甲基亚砜漂洗,抽干,合并产品的二甲基亚砜溶液。
[0042]
4)减压精馏产品,控制真空度在3mmhg,缓慢升温精馏,得产品:508.3g,纯度99.3%:收率:93%。
[0043]
实施例3
1)20l反应釜中加入15kg n,n-二甲苯胺,搅拌,温度控制在10-20℃,分批次加入3.1kg 三聚氯氰,加入完毕,搅拌6h,检测三聚氯氰已经反应完毕。
[0044]
2)配制1.9kg 1,6-己二醇和2kg的二氯甲烷的混合溶液,搅拌,将温度控制在-5-0℃,滴加1)中所配反应液,滴加完毕,0℃下搅拌反应2小时,停止制冷,自然升至25℃,取样中控,当1,6-己二醇完全反应,反应结束;3)将上述所得的混合反应液低真空抽滤,滤饼用500g的二氯甲烷漂洗,抽干,合并滤液。
[0045]
4)常压精馏回收二氯甲烷,回收二氯甲烷完毕后,控制真空度在3mmhg,缓慢升温精馏,得产品:2.06kg,纯度99.0%:收率:94%。
[0046]
对比例1(与实施例1相比,区别仅在于步骤1)中溶剂和三聚氯氰比例降低至2:1以下)1)20l反应釜中加入4kg n,n-二甲苯胺,搅拌,温度控制在10-20℃,分批次加入3.1kg 三聚氯氰,加入完毕,搅拌6h,检测三聚氯氰已经反应完毕。
[0047]
2)配制1.9kg 1,6-己二醇和2kg的二氯甲烷的混合溶液,搅拌,将温度控制在-5-0℃,滴加1)中所配反应液,滴加完毕,0℃下搅拌反应2小时,停止制冷,自然升至25℃,取样中控,当1,6-己二醇完全反应,反应结束;3)将上述所得的混合反应液低真空抽滤,滤饼用500g的二氯甲烷漂洗,抽干,合并滤液。
[0048]
4)常压精馏回收二氯甲烷,回收二氯甲烷完毕后,控制真空度在3mmhg,缓慢升温精馏,得产品:1.86kg,纯度97.2%:收率:85%。
[0049]
对比例2(与实施例2相比,区别仅在于步骤2)中控制温度在25-30℃之间)。
[0050]
1)四口瓶中加入3.7kg 二甲基亚砜,搅拌,温度控制在10-20℃,分批次加入738g三聚氯氰,加入完毕,搅拌2h,检测三聚氯氰已经反应完毕。
[0051]
2)配制473g 1,6-己二醇和700g的二甲基亚砜的混合溶液,搅拌,将温度控制在25-30℃,滴加1)中所配反应液,滴加完毕,10℃下搅拌反应2小时,停止制冷,自然升温至25℃,取样中控,当1,6-己二醇完全反应,反应结束。
[0052]
3)将上述所得的混合反应液低真空抽滤,滤饼用100g的二甲基亚砜漂洗,抽干,合并产品的二甲基亚砜溶液。
[0053]
4)减压精馏产品,控制真空度在3mmhg,缓慢升温精馏,得产品:453g,纯度97.6%:收率:83%。
[0054]
对比例3(与实施例2相比,区别仅在于三聚氯氰:己二醇=1.2:1)。
[0055]
1)四口瓶中加入3.7kg 二甲基亚砜,搅拌,温度控制在10-20℃,分批次加入885.6g三聚氯氰,加入完毕,搅拌2h,检测三聚氯氰已经反应完毕。
[0056]
2)配制473g 1,6-己二醇和700g的二甲基亚砜的混合溶液,搅拌,将温度控制在25-30℃,滴加1)中所配反应液,滴加完毕,10℃下搅拌反应2小时,停止制冷,自然升温至25℃,取样中控,当1,6-己二醇完全反应,反应结束。
[0057]
3)将上述所得的混合反应液低真空抽滤,滤饼用100g的二甲基亚砜漂洗,抽干,合并产品的二甲基亚砜溶液。
[0058]
4)减压精馏产品,控制真空度在3mmhg,缓慢升温精馏,得产品:431g,纯度97.6%:
收率:79%。
[0059]
对比例4(与实施例3相比,区别仅在于提高了真空度)。
[0060]
1)20l反应釜中加入15kg n,n-二甲苯胺,搅拌,温度控制在10-20℃,分批次加入3.1kg 三聚氯氰,加入完毕,搅拌6h,检测三聚氯氰已经反应完毕。
[0061]
2)配制1.9kg 1,6-己二醇和2kg的二氯甲烷的混合溶液,搅拌,将温度控制在-5-0℃,滴加1)中所配反应液,滴加完毕,0℃下搅拌反应2小时,停止制冷,自然升至25℃,取样中控,当1,6-己二醇完全反应,反应结束;3)将上述所得的混合反应液低真空抽滤,滤饼用500g的二氯甲烷漂洗,抽干,合并滤液。
[0062]
4)常压精馏回收二氯甲烷,回收二氯甲烷完毕后,控制真空度在1mmhg,缓慢升温精馏,得产品:1.97kg,纯度97.9%:收率:90%。
[0063]
通过各实施例与对比例的对比可知,本发明中各工艺参数均是非常重要的,不可简单调整、修改,否则极易导致产物纯度和收率的大幅降低。
[0064]
本发明中所用原料、设备,若无特别说明,均为本领域的常用原料、设备;本发明中所用方法,若无特别说明,均为本领域的常规方法。
[0065]
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips