
 商标分类
商标分类  商标转让
商标转让 
一种阿朴菲生物碱及其中间体的制备方法与流程
 2021-02-02 16:02:00|
2021-02-02 16:02:00| 407|
407| 起点商标网
起点商标网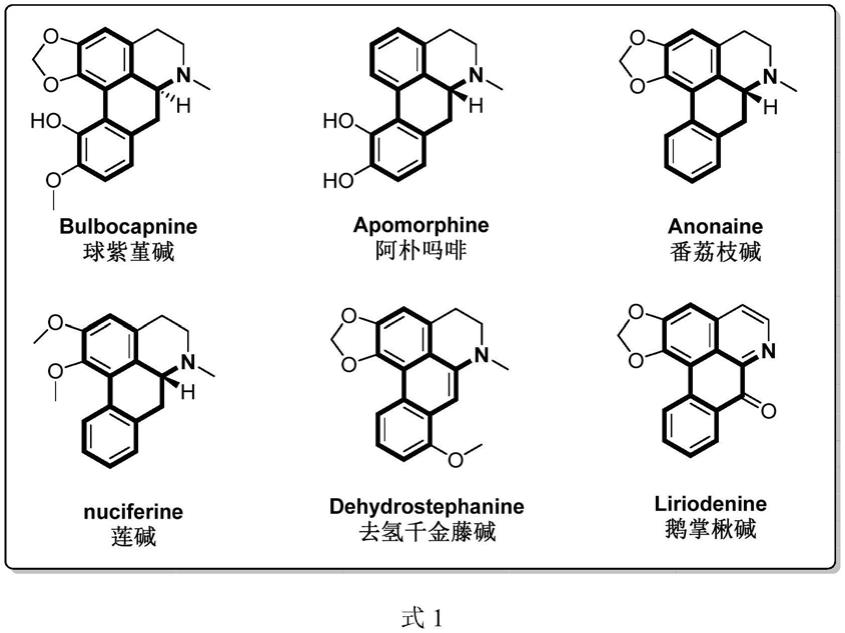
[0001]
本发明属于天然产物及其中间体制备领域,具体涉及一种阿朴菲生物碱及其中间体的制备方法。
背景技术:
[0002]
阿朴菲类生物碱是一类自然界中广泛分布的异喹啉生物碱(式1列出了几种常见的阿朴菲类生物碱)(天然产物研究与开发,2006,18(2):316-324)。研究表明,阿朴菲类生物碱具有良好的抗帕金森病(j.med.chem.2007,50,171-181)、抗肿瘤(anti-cancer.agent.me.2005,5,173-182)、抗病毒(nat.prod.res.2009,24,1395-1402)、抗脊髓灰白质病毒(j.nat.prod.1998,61,480-484)等生物活性。
[0003][0004]
阿朴菲类生物碱的母核为四氢二苯并[de,g]喹啉的四环骨架结构。传统合成该骨架结构的路线如下:第一步,以芳香乙胺和芳香乙酸作为起始原料通过缩合反应生成酰胺底物;第二步,酰胺底物通过bischler-napieralski分子内环化反应生成四氢喹啉底物;最后,四氢喹啉底物通过芳基碳碳键偶联反应实现四环骨架的构筑,生成目标产物。最后一步为该合成路线的关键步骤,不同的阿朴菲类生物碱由于环上连有不同的取代基需要采用不同的合成方法,主要包括经典的pschorr反应、酚的氧化偶联反应、光环化反应、自由基环化反应以及邻位直接芳基化反应,这些反应大大增加了合成策略的复杂性。
技术实现要素:
[0005]
本发明提供了一种阿朴菲生物碱及其中间体的制备方法,该制备方法通过直接在芳环特定位置引入乙氧羰甲基,然后再经过还原和关环等步骤,可以合成带各种官能团的阿朴菲生物碱及其衍生物。
[0006]
一种阿朴菲生物碱中间体的制备方法,包括:
[0007]
在钯催化剂和添加剂的作用下,n-芳基-喹啉-2-甲酰胺与溴乙酸乙酯发生烷基化反应,得到阿朴菲生物碱中间体;
[0008]
所述的n-芳基-喹啉-2-甲酰胺的结构如式(ii)所示:
[0009][0010]
所述的阿朴菲生物碱中间体的结构如式(iii)所示:
[0011][0012]
r选自h、c
1
~c
5
烷基或者c
1
~c
5
烷氧基,虚线表示该苯环可以存在,也可以不存在。
[0013]
作为优选,所述的钯催化剂为醋酸钯,所述的添加剂为phcook。
[0014]
作为优选,所述的烷基化反应的溶剂为1,2-二氯乙烷。
[0015]
作为优选,所述的烷基化反应的温度为80~100℃,反应时间为8~24小时。
[0016]
本发明还提供了一种阿朴菲生物碱的制备方法,包括以下步骤:
[0017]
(1)按照上述的制备方法得到阿朴菲生物碱中间体;
[0018]
(2)在还原剂的作用下,阿朴菲生物碱中间体发生还原反应,得到n-2-羟乙基芳基-喹啉-2-甲酰胺;
[0019]
所述的n-2-羟乙基芳基-喹啉-2-甲酰胺的结构如式(iv)所示:
[0020][0021]
(3)在碱和对甲苯磺酰氯的作用下,n-2-羟乙基芳基-喹啉-2-甲酰胺发生磺酰化反应,得到磺酰酯中间体;
[0022]
所述的磺酰酯中间体的结构如式(v)所示:
[0023][0024]
(4)在nah的作用下,磺酰酯中间体发生环化反应,得到2,3-二氢-1h-苯并[d]喹啉类化合物;
[0025]
所述的2,3-二氢-1h-苯并[d]喹啉类化合物的结构如式(vi)所示:
[0026][0027]
(5)2,3-二氢-1h-苯并[d]喹啉类化合物与碘甲烷发生甲基化反应,反应结束后,经过后处理得到所述的阿朴菲生物碱;
[0028]
所述的阿朴菲生物碱的结构如式(i)所示:
[0029][0030]
r选自h、c
1
~c
5
烷基或者c
1
~c
5
烷氧基。
[0031]
作为优选,步骤(2)中,所述的还原剂为lialh
4
。
[0032]
作为优选,步骤(3)中,所述的碱为三乙胺,添加剂为dmap。
[0033]
作为优选,步骤(5)中,甲基化反应在koh的作用下,于dmf中进行。
[0034]
具体反应路线如下:
[0035][0036]
同现有技术相比,本发明的有益效果体现在:
[0037]
本发明从9-氨基菲起始原料出发,分别通过8-碳氢键官能化和碳氮环化反应得到去氢阿朴菲类生物碱。相对于传统的合成策略,该策略有以下三方面的优势:一、起始原料商业可以得到;二、合成策略简便,碳氢键官能化策略使反应更加经济、高效;三、底物适用性强,芳环上的各种官能团对环的构筑并没有影响,可以实现各种阿朴菲类生物碱的合成。
具体实施方式
[0038]
实施例1
[0039]
将喹啉-2-甲酸(20mmol),萘-1-胺(20mmol,2.86g)和三乙胺(40mmol,5.6ml)溶解在ch
2
cl
2
(40ml)中,然后在0℃滴加pocl
3
(3.76ml)。反应混合物在0℃搅拌0.5h.然后反应在室温下搅拌2h直到萘-1-胺消耗完。反应结束之后,反应混合物冷却至0℃,缓慢加入冰水淬灭反应。收集有机相,然后用二氯甲烷(3
×
20ml)萃取水相,合并有机相,用饱和nahco
3
(2
×
40ml)水溶液洗涤,然后用mgso
4
干燥。减压旋干溶剂,粗产物用二氯甲烷/石油醚重结晶得到目标产物ii-1。
[0040]
反应式如下:
[0041][0042]
产物表征数据如下:
[0043]
n-(naphthalen-1-yl)quinoline-2-carboxamide(ii-1cas号:298193-67-6),产量:4.29g;收率:72%;粉红色固体;mp=146-147℃;
1
h nmr(400mhz,cdcl
3
)δ10.95(s,1h),8.43(d,j=8.4hz,2h),8.36(d,j=8.0hz,1h),8.25(d,j=8.4hz,1h),8.16(d,j=8.4hz,1h),7.91(d,j=8.0hz,2h),7.81(t,j=7.2hz,1h),7.71(d,j=8.4hz,1h),7.59(m,4h);
13
c nmr(100mhz,cdcl
3
)δ162.4,149.9,146.3,138.0,134.2,132.5,130.4,129.9,129.5,128.9,128.2,127.9,126.5,126.3,126.1,126.0,125.1,120.5,118.8,118.7;hrms(esi)calcd for c
20
h
14
n
2
o[m+h]
+ 299.1179,found 299.1182。
[0044]
实施例2制备原料n-(phenanthren-9-yl)quinoline-2-carboxamide ii-2
[0045]
反应式如下:
[0046][0047]
步骤1:
[0048]
向喹啉-2-甲酸(3.46g,20mmol,1equiv)和(boc)
2
o(5.68g,26mmol,1.3equiv)的1,4-二氧六环(100ml)溶液中,加入吡啶(2ml)。搅拌10min,分批加入nh
4
hco
3
(2.06g,26mmol,1.3equiv),然后在室温下搅拌24小时。反应完成后,旋干溶剂,残留物用etoac(200ml)溶解,有机相用nahco
3
(3
×
80ml)和水(80ml)洗涤,无水硫酸钠干燥,然后除去溶剂,干燥得到喹啉-2-甲酰胺。产物的结构和表征数据如下:
[0049][0050]
quinoline-2-carboxamide(cas no.5382-42-3):2.96g,86%收率;white solid;mp=126-128℃;
1
h nmr(400mhz,cdcl
3
)δ8.31(d,j=8.4hz,1h),8.25(d,j=8.4hz,1h),8.18(s,1h),8.08(d,j=8.4hz,1h),7.82(d,j=8.4hz,1h),7.72(t,j=7.6hz,1h),7.58(t,j=7.6hz,1h),6.92(s,1h);
13
c nmr(100mhz,cdcl
3
)δ167.5,149.5,146.6,137.4,130.1,129.8,129.3,128.0,127.7,118.8;
[0051]
步骤2:
[0052]
将喹啉-2-甲酰胺(1.72g,10.0mmol,1.0equiv)、菲-9-硼酸(4.44g,20.0mmol,2.0equiv),cu(oac)
2
(1.99g,11mmol,1.1equiv),和吡啶(1.58g,20.0mmol,2.0equiv)分散到ch
2
cl
2
(40ml)形成混合物,然后在室温下搅拌12小时。反应混合物在真空上进行浓缩,剩余物用硅胶柱(洗脱剂为二氯甲烷和石油醚)进行纯化以30%的得率得到目标产物。
[0053]
产物表征数据如下:
[0054]
n-(phenanthren-9-yl)quinoline-2-carboxamide(ii-2):1.04g,30%收率;黄色固体;mp=191-193℃;
1
h nmr(500mhz,cdcl
3
)δ10.96(s,1h),8.74(m,2h),8.61(m,1h),8.44(d,j=8.0hz,1h),8.36(d,j=8.5hz,1h),8.25(d,j=8.0hz,1h),8.21(d,j=8.5hz,1h),7.91(m,2h),7.81(t,j=8.0hz,1h),7.72(m,2h),7.61(m,3h);
13
c nmr(125mhz,cdcl
3
)δ162.6,149.9,146.3,138.0,132.1,131.2,130.4,130.3,129.9,129.6,128.8,128.4,128.2,127.9,127.1,127.0,126.8,126.6,126.1,123.6,122.4,121.0,118.8,118.5;hrms(esi)calcd for c
24
h
16
n
2
o[m+h]
+ 349.1335,found 349.1337.
[0055]
实施例3烷基化反应
[0056]
在空气氛围下,将酰胺ii-1或ii-2(0.25mmol,1.0equiv),溴乙酸乙酯(0.5mmol,84mg),pd(oac)
2
(0.025mmol,6mg),phcook(0.5mmol,80mg)和1,2-二氯乙烷(2.0ml)混合后,放置在35ml带有聚四氟乙烯帽的压力反应管中。反应管在90℃加热12h。反应混合物冷却至室温,用乙酸乙酯(5ml)稀释,硅藻土过滤,真空浓缩,剩余物用硅胶柱(洗脱剂为石油醚和乙酸乙酯)纯化,得到目标产物iii-1或iii-2。
[0057]
化合物iii-1的结构和表征数据如下:
[0058][0059]
ethyl 2-(8-quoinoline-2-carboxamido)naphthalen-1-yl)acetate(iii-1):83mg;收率:86%;柱层析得到白色固体(淋洗液:乙酸乙酯/石油醚=1/6,v/v);mp=128-129℃;
1
h nmr(400mhz,cdcl
3
)δ10.87(s,1h),8.44(d,j=8.0,1h),8.39(d,j=8.0hz,1h),8.15(d,j=8.0hz,1h),7.93(d,j=8.0hz,1h),7.83(m,4h),7.66(t,j=6.8hz,1h),7.55(t,j=7.4hz,1h),7.41(t,j=7.2hz,1h),7.34(d,j=6.4hz,1h),4.34(s,2h),4.11(q,j=6.8hz,2h),0.97(t,j=6.4hz,3h);
13
c nmr(100mhz,cdcl
3
)δ172.8,164.1,150.2,146.6,137.7,136.2,132.5,131.2,130.3,129.7,129.5,129.4,129.2,129.0,128.6,128.2,127.9,127.0,125.5,125.3,119.3,61.1,42.0,13.8.hrms(esi)calcd for c
24
h
20
n
2
o
3
[m+h]
+ 385.1547,found 385.1549.
[0060]
化合物iii-2的结构和表征数据如下:
[0061][0062]
ethyl 2-(10-(quinoline-2-carboxamido)phenanthren-1-yl)acetate(iii-2):78mg,72%收率;柱层析得到白色固体(淋洗液:乙酸乙酯/石油醚=1/4,v/v);mp=134-138℃;
1
h nmr(500mhz,cdcl
3
)δ10.81(s,1h),8.79(d,j=8.0,1h),8.68(d,j=8.0hz,1h),8.46(d,j=8.0,1h),8.40(d,j=9.0hz,1h),8.17(d,j=8.5hz,1h),8.13(s,1h),7.93(d,j=8.0hz,1h),7.89(d,j=7.0hz,1h),7.79(t,j=7.5hz,1h),7.63(m,4h),7.45(d,j=6.5hz,
1h),4.40(s,2h),4.08(q,j=7.0hz,2h),0.94(t,j=7.0hz,3h);
13
c nmr(125mhz,cdcl
3
)δ172.6,164.0,150.2,146.5,137.8,133.1,132.0,131.4,130.7,130.3,130.1,130.0,129.7,129.5,128.5,128.3,128.2,127.9,127.1,127.0,126.9,126.2,123.5,123.0,119.3,61.1,42.4,13.8;hrms(esi)calcd for c
28
h
22
n
2
o
3
[m+h]
+ 435.1703,found 435.1698.
[0063]
实施例4还原酯基制备iv-1和iv-2
[0064][0065]
在0℃、搅拌条件下,向化合物iii-1或iii-2(1.2mmol)的10ml无水四氢呋喃溶液中,在5分内分批加入lialh
4
(228mg,6.0mmol)。然后,在0℃继续反应1小时直到起始原料消耗完。然后,缓慢加入冰水(5ml)进行淬灭,然后将20%naoh(0.5ml)加入反应液,用etoac(3
×
10ml)进行萃取,合并的有机层水洗(10ml),无水硫酸钠干燥。旋干溶剂,剩余物用进行用乙酸乙酯/石油醚作为淋洗剂进行柱层析得到化合物iv-1或iv-2。
[0066]
化合物iv-1的结构表征数据如下:
[0067][0068]
n-(8-(2-hydroxyethyl)naphthalen-1-yl)quinoline-2-carboxamide(iv-1):267mg,65%收率;柱层析得到淡绿色固体(淋洗液:乙酸乙酯/石油醚=1/4,v/v);mp=141-143℃.
1
h nmr(600mhz,cd
3
cl)δ11.17(s,1h),8.48(d,j=9.0hz,1h),8.39(d,j=8.4hz,1h),8.21(d,j=8.4hz,1h),8.02(d,j=7.8hz,1h),7.93(d,j=7.8hz,1h),7.81(t,j=7.2hz,1h),7.78(m,2h),7.67(t,j=7.2hz,1h),7.52(t,j=7.8hz,1h),7.42(t,j=7.8hz,1h),7.36(d,j=6.6hz,1h),4.20(t,j=6.0hz,2h),3.60(t,j=6.0hz,2h),3.34(s,1h);
13
c nmr(150mhz,cd
3
cl)δ162.1,150.1,146.0,137.6,135.6,134.1,132.1,130.2,130.1,129.0,128.8,128.1,128.0,127.8,127.5,127.4,125.0,124.8,124.7,119.3,64.6,38.2;hrms(apci)calcd for c
22
h
18
n
2
o
2
[m+h]
+ 343.1441,found 343.1432.
[0069]
化合物iv-2的结构表征数据如下:
[0070][0071]
n-(8-(2-hydroxyethyl)phenanthren-9-yl)quinoline-2-carboxamide(iv-2):376mg,80%收率;柱层析得到白色固体(淋洗液:乙酸乙酯/石油醚=1/4,v/v);mp=150-153℃;
1
h nmr(500mhz,cd
3
cl)δ11.08(s,1h),8.65(d,j=8.5hz,1h),8.58(d,j=8.0hz,
1h),8.44(d,j=8.5hz,1h),8.32(d,j=8.5hz,1h),8.28(s,1h),8.16(d,j=8.5hz,1h),7.87(d,j=8.5hz,1h),7.84(d,j=8.5hz,1h),7.76(t,j=7.5hz,1h),7.58(m,4h),7.43(d,j=7.0hz,1h),4.19(t,j=6.0hz,2h),3.68(s,1h),3.59(t,j=6.0hz,2h);
13
c nmr(125mhz,cd
3
cl)δ162.5,150.6,146.4,138.1,135.3,132.8,131.4,131.3,130.6,129.6,129.4,129.1,128.3,128.2,127.9,127.8,126.9,126.5,126.3,124.9,122.9,122.4,119.8,64.9,38.8;hrms(esi)calcd for c
26
h
20
n
2
o
2
[m+h]
+ 393.1598,found 393.1591.
[0072]
实施例5环化反应制备vi-1或vi-2
[0073][0074]
(1)将化合物iv-1或iv-2(0.8mmol),dmap(0.16mmol,20mg)和et
3
n(6.4mmol,646mg)溶解在ch
2
cl
2
(5ml)中,然后在0℃滴加tscl(4.0mmol,763mg)。反应混合物在室温下搅拌3.0-4.0h,直到化合物iv-1或iv-2消耗完全。然后,加水淬灭反应。收集有机相,水相用ch
2
cl
2
(3
×
10ml)萃取。有机相用硫酸镁干燥,减压旋干溶剂,剩余物用柱层析进行纯化(淋洗剂,v-1:乙酸乙酯:石油醚=1:8,v-2:二氯甲烷:石油醚=1:1,v/v)得到化合物v-1(223mg,56%,黄色油状物)或v-2(197mg,45%,白色固体)。
[0075]
(2)将化合物v-1或v-2(0.4mmol)溶解在5ml无水thf中,在0℃、搅拌条件下缓慢加入nah(48mg,2.0mmol)。反应混合物在50℃反应12小时。然后,缓慢加水淬灭反应,用etoac(3
×
10ml)萃取,合并有机相,用无水硫酸镁干燥。在真空条件下,除去溶剂,剩余物用柱层析(用乙酸乙酯/石油醚作为淋洗剂)进行纯化,得到化合物vi-1或vi-2。
[0076]
化合物vi-1的结构和表征数据如下:
[0077][0078]
2,3-dihydro-1h-benzo[de]quinoline(vi-1):49mg,72%收率;柱层析后得到无色油状液体(洗脱剂,乙酸乙酯/石油醚=1/15,v/v);
1
h nmr(600mhz,cd
3
cl)δ7.67(d,j=8.4hz,1h),7.40(t,j=7.8hz,1h),7.31(t,j=7.8hz,1h),7.26(d,j=8.4hz,1h),7.14(d,j=6.6hz,1h),6.63(d,j=7.8hz,1h),4.13(s,1h),3.53(t,j=6.0hz,2h),3.26(t,j=6.0hz,2h);
13
c nmr(150mhz,cd
3
cl)δ144.3,134.6,133.1,126.7,126.0,125.8,122.1,121.7,117.3,106.9,41.8,30.7;hrms(esi)calcd for c
12
h
11
n[m+h]
+ 170.0964,found 170.0962.
[0079]
化合物vi-2的结构和表征数据如下:
[0080][0081]
5,6-dihydro-4h-dibenzo[de,g]quinoline(vi-2):75mg,86%收率;柱层析后得
到白色固体(洗脱剂,乙酸乙酯/石油醚=1/20,v/v);mp=127-128℃;
1
h nmr(500mhz,cd
3
cl)δ8.42(d,j=8.0hz,2h),7.52(d,j=7.5hz,1h),7.47(t,j=7.5hz,1h),7.38(d,j=8.0hz,1h),7.28(d,j=7.5hz,1h),7.23(d,j=6.0hz,1h),6.70(s,1h),4.10(s,1h),3.42(t,j=6.0hz,2h),3.18(t,j=6.0hz,2h).
13
c nmr(125mhz,cd
3
cl)δ141.4,133.5,133.4,131.2,126.9,126.5,126.0,125.6,124.8,122.8,122.7,121.0,104.1,41.5,30.7;hrms(esi)calcd for c
16
h
13
n[m+h]
+ 220.1121,found 220.1117.
[0082]
实施例6:甲基化制备i-1和i-2
[0083][0084]
将化合物vi-1或vi-2(0.25mmol)溶解在2ml dmf中,搅拌条件下加入koh(14mg,0.25mmol)。继续搅拌10分钟后,滴加碘甲烷(178mg,1.25mmol)。反应体系在室温下反应2~3小时,反应完成后,反应液用水稀释,然后用etoac(3
×
5ml)萃取,有机相用水(10ml)洗涤,无水硫酸钠干燥,减压旋干溶剂,剩余物以乙酸乙酯/石油醚作为淋洗剂用柱层析进行纯化,得到化合物i-1或i-2。
[0085]
化合物i-1的结构和表征数据如下:
[0086][0087]
1-methyl-2,3-dihydro-1h-benzo[de]quinoline(i-1):28mg,收率:62%;柱层析后得到无色油状物质(洗脱剂:乙酸乙酯/石油醚=1/20,v/v);
1
h nmr(600mhz,cd
3
cl)δ7.64(d,j=8.4hz,1h),7.37(t,j=7.8hz,2h),7.24(t,j=8.4hz,1h),7.12(d,j=7.2hz,1h),6.61(s,1h),3.41(t,j=6.0hz,2h),3.28(t,j=6.0hz,2h),3.04(s,3h);
13
c nmr(150mhz,cd
3
cl)δ145.9,134.4,132.8,126.8,125.8,125.7,122.4,122.2,117.1,104.6,50.9,40.3,30.7;hrms(apci)calcd for c
13
h
13
n[m+h]
+ 184.1121,found 184.1115.
[0088]
化合物i-2的结构和表征数据如下:
[0089][0090]
6-methyl-5,6-dihydro-4h-dibenzo[de,g]quinoline(i-2,cas号:7630-71-9):42mg,72%收率;柱层析后的得到无色油状物质(洗脱剂,乙酸乙酯/石油醚=1/40,v/v);
1
h nmr(500mhz,cd
3
cl)δ8.43(d,j=8.5hz,2h),7.62(d,j=8.0hz,1h),7.47(t,j=7.5hz,1h),7.39(t,j=7.5hz,1h),7.29(t,j=7.5hz,1h),7.23(d,j=7.0hz,1h),6.67(s,1h),
3.33(t,j=6.0hz,2h),3.23(t,j=6.0hz,2h),3.03(s,3h);
13
c nmr(125mhz,cd
3
cl)δ143.5,133.8,133.2,131.0,126.9,126.7,126.2,125.2,124.8,123.4,122.8,122.6,121.0,102.3,50.6,40.4,30.7;hrms(esi)calcd for c
17
h
15
n[m+h]
+ 234.1277,found 234.1271。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips