
 商标分类
商标分类  商标转让
商标转让 
一种有机化合物及其应用及采用该化合物的有机电致发光器件的制作方法
 2021-02-02 15:02:14|
2021-02-02 15:02:14| 415|
415| 起点商标网
起点商标网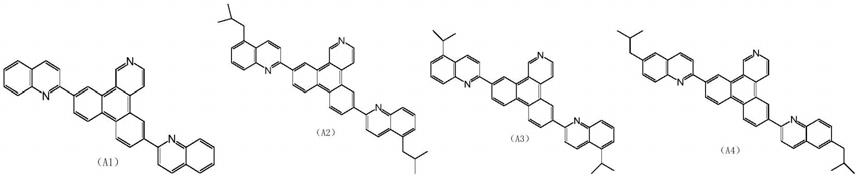
[0001]
本发明涉及一种新型有机化合物,尤其涉及一种用于有机电致发光器件的化合物及采用该类化合 物的有机电致发光器件。
背景技术:
[0002]
有机电致发光显示器(以下简称oled)具有自主发光、低电压直流驱动、全固化、视角宽、重 量轻、组成和工艺简单等一系列的优点,与液晶显示器相比,有机电致发光显示器不需要背光源,视 角大,功率低,其响应速度可达液晶显示器的1000倍,其制造成本却低于同等分辨率的液晶显示器, 因此,有机电致发光器件具有广阔的应用前景。
[0003]
随着oled在照明和显示两大领域的不断推进,人们对于其核心材料的研究也更加关注。这是因 为一个效率好、寿命长的oled器件通常是器件结构以及各种有机材料的优化搭配的结果,这就为化 学家们设计开发各种结构的功能化材料提供了极大的机遇和挑战。常见的功能化有机材料有:空穴注 入材料、空穴传输材料、空穴阻挡材料、电子注入材料、电子传输材料,电子阻挡材料以及发光主体 材料和发光客体(染料)等。
[0004]
为了制备驱动电压更低、发光效率更好、器件使用寿命更长的oled发光器件,实现oled器件 的性能不断提升,不仅需要对oled器件结构和制作工艺进行创新,更需要对oled器件中的光电功 能材料不断研究和创新,以制备出具有更高性能的功能材料。基于此,oled材料界一直致力于开发 新的有机电致发光材料以实现器件低启动电压、高发光效率和更优的使用寿命。
[0005]
电子传输材料以传输电子为主,负责将电子载流子由金属阴极传递并注入发光层,电子传输材料 的性能对oled器件的效率具有较大的影响。能够对oled器件的效率具有较大提升作用的电子传输 材料,通常具有以下特性:(1)材料的电化学还原性可逆,这是由于在有机薄膜中电子的传导过程是 一系列的氧化还原过程;(2)材料的homo和lumo能级合适,使电子的注入势垒最小;(3)材料 的电子迁移率高;(4)材料的玻璃化温度(tg)和热分解稳定性较高,避免工作中产生的焦耳热对器 件效率和寿命的影响;目前专利文献中公开可用于oled器件的电子传输材料主要包括含氮五元杂环 类例如噁二唑,三氮唑,咪唑,噁唑,噻唑,三嗪类,喹啉类,吡啶类,邻二氮杂菲类,有机硼类等。
[0006]
随着oled产品逐步进入市场,人们对这类产品的性能有越来越高的要求。当前使用的oled材 料和器件结构无法完全解决oled产品效率、寿命、成本等各方面的问题。
[0007]
然而,为了进一步满足对oled器件的光电性能不断提升的需求,以及移动化电子器件对于节能 的需求,需要不断地开发新型的、高效的oled材料,其中开发新的具有高电子注入能力和高迁移率 的电子传输材料具有很重要的意义。
技术实现要素:
[0008]
本发明提供一种通式化合物,该化合物具有如下式(1)所示的结构:
[0009][0010]
式(1)中,ar
1
和ar
2
分别独立地选自取代或未取代的c6-c30的芳基、取代或未取代的c3-c30的缺 电子杂芳基中的一种,且ar
1
和ar
2
中至少有一个选自缺电子杂芳基。
[0011]
进一步优选的,ar
1
和ar
2
中均选自取代或未取代的c3-c30的缺电子杂芳基。
[0012]
再进一步的,所述的缺电子杂芳基优选选自三嗪基、嘧啶基、苯并嘧啶基、吡啶基、联吡啶基、苯并吡 啶基、二氮杂萘基、二氮杂菲基、吡嗪基、喹啉基、异喹啉基、喹唑啉基、喹喔啉基、邻菲罗啉基或哒嗪基 中的一种。
[0013]
具体说,上述的“缺电子杂芳基”是指该基团取代含杂原子(优选杂原子为氮)的苯环上的氢后,该苯环 上的电子云密度降低的基团,通常这样的基团的哈米特值大于0.6。所述哈米特值是指对特定基团电荷亲和 力的表征,是吸电子基团(正哈米特值)或给电子基团(负哈米特值)的度量。在thomas h.lowry和katheleen schueller richardson,“mechanism and theory in organic chemistry
’
,new york,1987,143-151页中更详细描述 了哈米特方程,此处引作参考。
[0014]
更进一步优选的,所述ar
1
和ar
2
分别独立地选自取代或未取代的下述基团中的一种:苯基、萘基、联 苯基、三联苯基、吡啶基、联吡啶基、苯并吡啶基、苯并嘧啶基、二氮杂萘基、二氮杂菲基、喹啉基、异喹 啉基、喹唑啉基、三嗪基、吡嗪基、哒嗪基、苯并咪唑基、嘧啶基或邻菲罗啉基。
[0015]
当上述基团存在取代基时,所述取代基选自卤素、氰基、羰基、c1-c10的烷基、c3-c10的环烷基、c2
-ꢀ
c10烯基、c1-c6的烷氧基或硫代烷氧基、c6-c30的单环芳烃或稠环芳烃基团、c3-c30的单环杂芳烃或稠 环杂芳烃基团中的一种或者至少两种的组合。
[0016]
作为上述取代或未取代的c6-c30芳基,优选具有6-20个骨架碳原子,优选所述芳基为由苯基、 联苯基、三联苯基、萘基、蒽基、菲基、茚基、芴基及其衍生物、荧蒽基、三亚苯基、芘基、苝基、 基和并四苯基所组成的组中的基团。所述联苯基选自2-联苯基、3-联苯基和4-联苯基;所述三联苯 基包括对-三联苯基-4-基、对-三联苯基-3-基、对-三联苯基-2-基、间-三联苯基-4-基、间-三联苯基-3-基 和间-三联苯基-2-基;所述萘基为1-萘基或2-萘基;所述蒽基选自由1-蒽基、2-蒽基和9-蒽基所组成 的组中;所述芴基选自由1-芴基、2-芴基、3-芴基、4-芴基和9-芴基所组成的组中;所述芴基衍生物 选自由9,9
’-
二甲基芴,9,9
’-
螺二芴和苯并芴所组成的组中;所述芘基选自由1-芘基、2-芘基和4-芘基 所组成的组中;所述并四苯基选自由1-并四苯基、2-并四苯基和9-并四苯基所组成的组中。
[0017]
作为本发明涉及的化合物的优选结构,可以举出以下a1~a50所示结构的化合物,但不限于这 些化合物:
[0018]
[0019]
[0020][0021]
作为本发明的另一个方面,还提供了一种如上所述的化合物在有机电致发光器件中的应用。具体说,优 选为在有机电致发光器件中作为电子传输层材料。
[0022]
作为本发明的又一个方面,还提供了一种有机电致发光器件,包括第一电极、第二电极和插入在 所述第一电极和第二电极之间的有机层,其特征在于,所述有机层中含有如上所述的通式(1)的化合 物,或者含有如上所述的a1~a50所示结构的化合物。
[0023]
上述本发明化合物作为电子传输层材料性能优异的具体原因尚不明确,推测可能是以下的原因:
[0024]
本发明所制备的化合物至少含有一个缺电子杂芳基取代基团,优选含有两个缺电子杂芳基取代基团,通 过不同的合成工艺构建新分子结构与现有技术常用的单个吡啶、喹啉、菲啰啉、三嗪、咪唑、噻唑或嘧啶等 结构相比,本发明所制备的化合物具有更大的共轭共平面结构,含有足够的拉电子基团,产品的缺电子性更 好,有利于电子的流动,提高产品的迁移率,所以本发明所制备的电子传输材料可以有效地提高器件中电子 注入与迁移率,从而确保器件获得高发光效率和低启动电压。
具体实施方式
[0025]
为了使本领域技术人员更好地理解本发明,下面结合具体实施方式对本发明作进
一步详细说明。
[0026]
本发明中未提到的合成方法的化合物的都是通过商业途径获得的原料产品。实施例中所用的各种化学 药品如石油醚、乙酸乙酯、正己烷、甲苯、四氢呋喃、二氯甲烷、四氯化碳、丙酮、碳酸铯、磷酸钾、叔丁 醇钠、pd(dppf)cl
2
、pd(pph3)
4
、pd(oac)
2
、醋酸钯、三环己基膦、s-phos双联频哪醇硼酸酯、4-吡 啶硼酸、2-溴-5-氯碘苯、2,4-二氯碘苯、2-溴喹啉、2-溴异喹啉、2-溴-5-异丙基喹啉、2-溴-6-异丁基喹 啉、4,6-二苯基-2-氯嘧啶、4,6-二苯基-2-氯三嗪、5-溴-1,10-菲啰啉,中间体m1,m2,m5,m6,m9从市场 上进行定制等
[0027]
本发明中的中间体和化合物的分析检测使用absciex质谱仪(4000qtrap)和布鲁克核磁共振仪(400m)。
[0028]
化合物的合成实施例:
[0029]
主要中间体的合成
[0030]
合成实施例1:
[0031]
化合物a1的合成:
[0032][0033]
中间体m1直接从市场上进行定制合成。
[0034]
中间体m3的合成:
[0035]
3l四口瓶中加入中间体m1(89.5g,0.3mol),双联频哪醇硼酸酯(182.9g,0.72mol),pd2(dba) 3(5.5g),s-phos(4.9g),磷酸钾(305.6g,1.44mol),二氧六环(1800ml),氮气置换两次,升温 到回流反应。随着反应时间的延长,体系颜色加深,反应6h,tlc监控反应完成。停止反应降至室温, 减压浓缩除去体系中的二氧六环,加入乙酸乙酯和水进行分液,有机相干燥。浓缩,过柱,正己烷煮 洗得到类白色固体75.2g,收率52.1%。
[0036]
化合物a1的合成:
[0037]
2l四口瓶中加入中间体m3(48.1g,0.1mol),2-溴喹啉(45.7g,0.22mol),甲苯(500ml),水 (100ml),碳酸钾(60.5g,0.44mol),开启搅拌,氮气置换两遍,升温到回流反应。反应12h,tlc 监控无原料剩余,停止反应自然降至室温。分液,水相用100ml乙酸乙酯萃取一遍,合并有机相,用 饱和食盐水100ml洗涤有机相两遍。有机相用25g无水硫酸钠进行干燥。过滤,浓缩,干法柱层析得 到类白色固体39.1g,收率80.9%。m/z理论值:483.57,m/z实测值:483.54。c35h21n3计算值: c,86.93;h,4.38;n,8.69,实测值:c,86.90;h,4.37;n,8.73。
[0038]
合成实施例2:
[0039]
化合物a2的合成:
[0040][0041]
2l四口瓶中加入中间体m3(48.1g,0.1mol),2-溴-5-异丁基喹啉(58.1g,0.22mol),甲苯(500ml), 水(100ml),碳酸钾(60.5g,0.44mol),开启搅拌,氮气置换两遍,升温到回流反应。反应10h,tlc 监控无原料剩余,停止反应自然降至室温。分液,水相用100ml乙酸乙酯萃取一遍,合并有机相,用 饱和食盐水100ml洗涤有机相两遍。有机相用25g无水硫酸钠进行干燥。过滤,浓缩,干法柱层析得 到类白色固体36.8g,收率61.7%。m/z理论值:595.79,m/z实测值:595.75。c43h37n3计算值: c,86.69;h,6.26;n,7.05,实测值:c,86.67;h,6.27;n,7.06。
[0042]
合成实施例3:
[0043]
化合物a3的合成:
[0044]
化合物a3与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-溴-5
-ꢀ
异丙基喹啉。中间体m3用量为(0.1mol),后处理得到类白色固体42.5g,收率71.3%。
[0045]
合成实施例4:
[0046]
化合物a4的合成:
[0047]
化合物a4与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-溴-6
-ꢀ
异丁基喹啉。中间体m3用量为(0.1mol),后处理得到类白色固体42.5g,收率71.3%。
[0048]
合成实施例5:
[0049]
化合物a5的合成:
[0050]
化合物a5与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为4,6-二苯 基-2-氯三嗪。中间体m3用量为(0.1mol),后处理得到类白色固体47.4g,收率68.5%。
[0051]
合成实施例6:
[0052]
化合物a6的合成:
[0053][0054]
2l四口瓶中加入中间体m3(48.1g,0.1mol),2-(4-溴苯基)-4,6-二苯基-1,3,5-三嗪(85.4g, 0.22mol),甲苯(700ml),水(100ml),碳酸钾(60.5g,0.44mol),开启搅拌,氮气
置换两遍,升温 到回流反应。反应11h,tlc监控无原料剩余,停止反应自然降至室温。分液,水相用150ml乙酸乙酯 萃取一遍,合并有机相,用饱和食盐水100ml洗涤有机相两遍。有机相用30g无水硫酸钠进行干燥。 过滤,浓缩,干法柱层析得到类白色固体51.8g,收率61.4%。m/z理论值:843.99,m/z实测值:843.97。 c59h37n7计算值:c,83.96;h,4.42;n,11.62,实测值:c,83.94;h,4.41;n,11.65。
[0055]
合成实施例7:
[0056]
化合物a7的合成:
[0057]
化合物a7与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-氯-4
-ꢀ
(2-萘基)-6-苯基-1,3,5-三嗪。中间体m3用量为(0.1mol),后处理得到类白色固体50.1g,收 率63.3%。
[0058]
合成实施例8:
[0059]
化合物a8的合成:
[0060]
化合物a8与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为4,4-(5
-ꢀ
氯-1,3-苯基)二吡啶。中间体m3用量为(0.1mol),后处理得到类白色固体48.6g,收率70.5%。
[0061]
合成实施例9:
[0062]
化合物a9的合成:
[0063]
化合物a9与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-氯-4
-ꢀ
苯基喹唑林。中间体m3用量为(0.1mol),后处理得到类白色固体36.8g,收率61.7%。
[0064]
合成实施例10:
[0065]
化合物a10的合成:
[0066]
化合物a10与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-溴异 喹啉。中间体m3用量为(0.1mol),后处理得到类白色固体31.3g,收率64.8%。
[0067]
合成实施例11:
[0068]
化合物a11的合成:
[0069]
化合物a11与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为1-溴-6
-ꢀ
异丁基异喹啉。中间体m3用量为(0.1mol),后处理得到类白色固体44.6g,收率74.9%。
[0070]
合成实施例12:
[0071]
化合物a12的合成:
[0072]
化合物a12与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为5-氯
-ꢀ
1,10-菲啰啉。中间体m3用量为(0.1mol),后处理得到类白色固体42.4g,收率72.4%。
[0073]
合成实施例13:
[0074]
化合物a13的合成:
[0075]
化合物a2与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为3-氯-1, 10-菲啰啉。中间体m3用量为(0.1mol),后处理得到类白色固体41.4g,收率70.6%。
[0076]
合成实施例14:
[0077]
化合物a14的合成:
[0078]
化合物a14与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-氯
-ꢀ
1,10-菲啰啉。中间体m3用量为(0.1mol),后处理得到类白色固体39.2g,收率66.9%。
[0079]
合成实施例15:
[0080]
化合物a15的合成:
[0081]
化合物a15与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为4-氯
-ꢀ
1,10-菲啰啉。中间体m3用量为(0.1mol),后处理得到类白色固体40.2g,收率68.7%。
[0082]
合成实施例16:
[0083]
化合物a16的合成:
[0084]
化合物a16与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为4,6-二 苯基-2-氯嘧啶。中间体m3用量为(0.1mol),后处理得到类白色固体41.8g,收率60.6%。
[0085]
合成实施例17:
[0086]
化合物a17的合成:
[0087]
化合物a17与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-(4
-ꢀ
氯苯基)-3-苯基吡嗪。中间体m3用量为(0.1mol),后处理得到类白色固体42.7g,收率61.9%。
[0088]
合成实施例18:
[0089]
化合物a18的合成:
[0090]
化合物a18与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-(4
-ꢀ
溴苯基)-4,6-二苯基嘧啶。中间体m3用量为(0.1mol),后处理得到类白色固体49.3g,收率58.6%。 合成实施例19:
[0091]
化合物a19的合成:
[0092]
化合物a19与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为5-氯
-ꢀ
2,2-联吡啶。中间体m3用量为(0.1mol),后处理得到类白色固体38.5g,收率71.6%。
[0093]
合成实施例20:
[0094]
化合物a20的合成:
[0095][0096]
2l四口瓶中加入中间体m3(48.1g,0.1mol),6-氯苯并-1,10-菲啰啉(58.2g,0.22mol),甲苯 (700ml),水(100ml),碳酸钾(60.5g,0.44mol),开启搅拌,氮气置换两遍,升温到回流反应。反 应11h,tlc监控无原料剩余,停止反应自然降至室温。分液,水相用
150ml乙酸乙酯萃取一遍,合并 有机相,用饱和食盐水120ml洗涤有机相两遍。有机相用30g无水硫酸钠进行干燥。过滤,浓缩,干 法柱层析得到类白色固体51.7g,收率75.4%。m/z理论值:685.79,m/z实测值:685.77。c49h27n5 计算值:c,85.82;h,3.97;n,10.21,实测值:c,85.84;h,3.95;n,10.21。
[0097]
合成实施例21:
[0098]
化合物a21的合成:
[0099]
化合物a21与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为1-(4
-ꢀ
氯苯基)-2-苯基苯并咪唑。中间体m3用量为(0.1mol),后处理得到类白色固体54.8g,收率71.6%。
[0100]
合成实施例22:
[0101]
化合物a22的合成:
[0102]
化合物a22与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-(4
-ꢀ
氯苯基)-1-苯基苯并咪唑。中间体m3用量为(0.1mol),后处理得到类白色固体57.1g,收率74.6%。
[0103]
合成实施例23:
[0104]
化合物a23的合成:
[0105]
化合物a23与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为2-(4
-ꢀ
氯苯基)-4-苯基喹唑林。中间体m3用量为(0.1mol),后处理得到类白色固体54.2g,收率68.6%。
[0106]
合成实施例24:
[0107]
化合物a24的合成:
[0108]
化合物a24与化合物a1合成方法相同,不同之处在于将化合物a1中的2-溴喹啉替换为6-氯
-ꢀ
2,3-二苯基喹喔啉。中间体m3用量为(0.1mol),后处理得到类白色固体52.2g,收率66.1%。
[0109]
合成实施例25:
[0110]
化合物a38的合成:
[0111][0112]
2l四口瓶中加入中间体m3(34.2g,0.1mol),联吡啶硼酸酯(31.1g,0.11mol),甲苯(700ml), 水(100ml),碳酸钾(30.4g,0.22mol),开启搅拌,氮气置换两遍,升温到回流反应。反应6h,tlc 监控无原料剩余,停止反应自然降至室温。分液,水相用200ml乙酸乙酯萃取一遍,合并有机相,用 饱和食盐水100ml洗涤有机相两遍。有机相用35g无水硫酸钠进行干燥。过滤,浓缩,干法柱层析得 到类白色固体34.2g,收率81.8%。
[0113]
2l四口瓶中加入中间体m4(34.2g,0.82mol),5-苯基喹啉-3-硼酸酯(33.1g,0.1mol),甲苯 (700ml),水(100ml),碳酸钾(23.5g,0.17mol),开启搅拌,氮气置换两遍,升温到回流反应。反 应6h,tlc监控无原料剩余,停止反应自然降至室温。分液,水相用200ml乙酸乙酯萃取一遍,合并 有机相,用饱和食盐水100ml洗涤有机相两遍。有机相用35g无水硫酸钠进行干燥。过滤,浓缩,干 法柱层析得到类白色固体37.2g,收率77.3%。m/z理论值:586.70,m/z实测值:586.68。c42h26n4 计算值:c,85.98;h,4.47;n,9.55,实测值:c,85.99;h,4.48;n,9.53。
[0114]
合成实施例26:
[0115]
化合物a41的合成:
[0116][0117]
2l四口瓶中加入中间体m2(34.2g,0.1mol),m6(43.6g,0.11mol),甲苯(700ml),水(100ml), 碳酸钾(30.4g,0.22mol),开启搅拌,氮气置换两遍,升温到回流反应。反应8h,tlc监控无原料剩 余,停止反应自然降至室温。分液,水相用200ml乙酸乙酯萃取一遍,合并有机相,用饱和食盐水100ml 洗涤有机相两遍。有机相用35g无水硫酸钠进行干燥。过滤,浓缩,干法柱层析得到类白色固体43.6g, 收率81.9%。
[0118]
2l四口瓶中加入中间体m7(43.6g,0.082mol),双联频哪醇硼酸酯(22.8g,0.9mol),二氧六环 (800ml),醋酸钾(15.7g,0.16mol),pd(dppf)cl2(0.6g)开启搅拌,氮气置换两遍,升温到90℃ 反应。反应8h,tlc监控无原料剩余,停止反应自然降至室温。分液,水相用250ml乙酸乙酯萃取一 遍,合并有机相,用饱和食盐水100ml洗涤有机相两遍。有机相用50g无水硫酸钠进行干燥。过滤, 浓缩,干法柱层析得到类白色固体37.6g,收率73.5%。
[0119]
2l四口瓶中加入中间体m8(37.6g,0.06mol),m9(16.5g,0.072mol),甲苯(400ml),水(100ml), 碳酸钾(19.3g,0.14mol),开启搅拌,氮气置换两遍,升温到回流反应。反应5h,tlc监控无原料剩 余,停止反应自然降至室温。分液,水相用300ml乙酸乙酯萃取一遍,合并有机相,用饱和食盐水150ml 洗涤有机相两遍。有机相用50g无水硫酸钠进行干燥。过滤,浓缩,干法柱层析得到类白色固体35.9g, 收率86.9%。m/z理论值:689.82,m/z实测值:689.80。c49h31n5计算值:c,85.32;h,4.53;n, 10.15,实测值:c,85.30;h,4.50;n,10.20。
[0120]
上述各个合成实施例所制备的本发明的化合物a1-a43分子式、分子量及收率详见下表1:
[0121]
表1:
[0122]
[0123]
[0124][0125]
器件实施例
[0126]
oled包括位于第一电极和第二电极,以及位于电极之间的有机材料层。该有机材料又可以分为多 个区域。比如,该有机材料层可以包括空穴传输区、发光层、电子传输区。
[0127]
在具体实施例中,在第一电极下方或者第二电极上方可以使用基板。基板均为具有机械强度、热 稳定性、防水性、透明度优异的玻璃或聚合物材料。此外,作为显示器用的基板上也可以带有薄膜晶 体管(tft)。
[0128]
第一电极可以通过在基板上溅射或者沉积用作第一电极的材料的方式来形成。当第一电极作为阳 极时,可以采用铟锡氧(ito)、铟锌氧(izo)、二氧化锡(sno2)、氧化锌(zno)等氧化物透明导电材料和 它们的任意组合。第一电极作为阴极时,可以采用镁(mg)、银(ag)、铝(al)、铝-锂(al-li)、钙(ca)、 镁-铟(mg-in)、镁-银(mg-ag)等金属或合金以及它们之间的任意组合。
[0129]
有机材料层可以通过真空热蒸镀、旋转涂敷、打印等方法形成于电极之上。用作有机材料层的化 合物可以为有机小分子、有机大分子和聚合物,以及它们的组合。
[0130]
空穴传输区位于阳极和发光层之间。空穴传输区可以为单层结构的空穴传输层(htl),包括只含 有一种化合物的单层空穴传输层和含有多种化合物的单层空穴传输层。空穴传输区也可以为包括空穴 注入层(hil)、空穴传输层(htl)、电子阻挡层(ebl)中的至少一层的多层结构。
[0131]
空穴传输区的材料可以选自、但不限于酞菁衍生物如cupc、导电聚合物或含导电掺杂剂的聚合物 如聚苯撑乙烯、聚苯胺/十二烷基苯磺酸(pani/dbsa)、聚(3,4-乙撑二氧
噻吩)/聚(4-苯乙烯磺酸盐) (pedot/pss)、聚苯胺/樟脑磺酸(pani/csa)、聚苯胺/聚(4-苯乙烯磺酸盐)(pani/pss)、芳香胺衍生 物如下面ht-1至ht-34所示的化合物;或者其任意组合。
[0132]
[0133][0134]
空穴注入层位于阳极和空穴传输层之间。空穴注入层可以是单一化合物材料,也
可以是多种化合 物的组合。例如,空穴注入层可以采用上述ht-1至ht-34的一种或多种化合物,或者采用下述hi1
-ꢀ
hi3中的一种或多种化合物;也可以采用ht-1至ht-34的一种或多种化合物掺杂下述hi1-hi3中的一 种或多种化合物。
[0135][0136]
发光层包括可以发射不同波长光谱的的发光染料(即掺杂剂,dopant),还可以同时包括主体材料 (host)。发光层可以是发射红、绿、蓝等单一颜色的单色发光层。多种不同颜色的单色发光层可以按 照像素图形进行平面排列,也可以堆叠在一起而形成彩色发光层。当不同颜色的发光层堆叠在一起时, 它们可以彼此隔开,也可以彼此相连。发光层也可以是能同时发射红、绿、蓝等不同颜色的单一彩色 发光层。
[0137]
根据不同的技术,发光层材料可以采用荧光电致发光材料、磷光电致发光材料、热活化延迟荧光 发光材料等不同的材料。在一个oled器件中,可以采用单一的发光技术,也可以采用多种不同的发光 技术的组合。这些按技术分类的不同发光材料可以发射同种颜色的光,也可以发射不同种颜色的光。
[0138]
在本发明的一方面,发光层采用荧光电致发光的技术。其发光层荧光主体材料可以选自、但不限 于以下所罗列的bfh-1至bfh-16的一种或多种的组合。
[0139]
[0140][0141]
在本发明的一方面,发光层采用荧光电致发光的技术。其发光层荧光掺杂剂可以选自、但不限于 以下所罗列的bfd-1至bfd-12的一种或多种的组合。
[0142]
[0143][0144]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层主体材料选自、但不限于gph-1 至gph-80中的一种或多种的组合。
[0145]
[0146]
[0147][0148]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层磷光掺杂剂可以选自、但不限于 以下所罗列的gpd-1至gpd-47的一种或多种的组合。
[0149]
[0150]
[0151][0152]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层磷光掺杂剂可以选自、但不限于 以下所罗列的rpd-1至rpd-28的一种或多种的组合。
[0153]
[0154][0155]
在本发明的一方面,发光层采用磷光电致发光的技术。其发光层磷光掺杂剂可以选自、但不限于 以下所罗列的ypd-1—ypd-11的一种或多种的组合。
[0156][0157]
oled有机材料层还可以包括发光层与阴极之间的电子传输区。电子传输区可以为单层结构的电子 传输层(etl),包括只含有一种化合物的单层电子传输层和含有多种化合物的单层电子传输层。电子 传输区也可以为包括电子注入层(eil)、电子传输层(etl)、空穴阻挡层(hbl)中的至少一层的多层结 构。
[0158]
本发明的一方面,电子传输层材料可以选自、但不限于以下所罗列的et-1至et-40的一种或多 种的组合。
[0159]
[0160]
[0161]
[0162][0163]
器件中还可以包括位于电子传输层与阴极之间的电子注入层,电子注入层材料包括但不限于以下 罗列的一种或多种的组合。
[0164]
liq,lif,nacl,csf,li
2
o,cs
2
co
3
,bao,na,li,ca。
[0165]
本发明的实施例中有机电致发光器件制备过程如下:
[0166]
将涂布了ito透明导电层的玻璃板在商用清洗剂中超声处理,在去离子水中冲洗,在丙酮:乙醇 混合溶剂中超声除油,在洁净环境下烘烤至完全除去水份,用紫外光和臭氧清洗,并用低能阳离子束 轰击表面;
[0167]
把上述带有阳极的玻璃基片置于真空腔内,抽真空至压强小于10-5
pa,在上述阳极层膜上利用多 源共蒸的方法,调节空穴传输材料ht-4蒸镀速率为0.1nm/s,空穴注入材料hi-3蒸镀速率7%比例 设定,蒸镀总膜厚为10nm;
[0168]
在空穴注入层之上真空蒸镀ht-4作为器件的第一空穴传输层,蒸镀速率为0.1nm/s,蒸镀总膜 厚为40nm;
[0169]
在第一空穴传输层之上真空蒸镀ht-14作为器件的第二空穴传输层,蒸镀速率为0.1nm/s,蒸镀 总膜厚为10nm;
[0170]
在第二空穴传输层之上真空蒸镀器件的发光层,发光层包括主体材料和染料材料,利用多源共蒸 的方法,调节主体材料bfh-4蒸镀速率为0.1nm/s,染料bfd-4蒸镀速率5%比例设定,蒸镀总膜厚 为20nm;
[0171]
在发光层之上真空蒸镀et-17作为器件的空穴阻挡层,蒸镀速率为0.1nm/s,蒸镀总膜厚为5 nm;
[0172]
在空穴阻挡层之上利用多源共蒸的方法制备电子传输层,电子传输材料可以选用本发明公开的 化合物a1~a50中的代表性化合物,或电子传输材料可以选用现有技术中的化合物l1~l2作为对比 材料,调节所选用电子传输材料的蒸镀速率为0.1nm/s,与et-57蒸镀速率100%比例设定,蒸镀总 膜厚为23nm;
[0173]
在电子传输层(etl)上真空蒸镀厚度为1nm的lif作为电子注入层,厚度为80nm的al层作为 器件的阴极。
[0174]
本发明选用的现有技术中电子传输材料包括以下化合物l1~l6:
[0175][0176]
对由上述过程制备的有机电致发光器件进行如下性能测定:
[0177]
在同样亮度下,使用photo research公司的pr 750型光辐射计st-86la型亮度计(北京师范 大学光电仪器厂)及keithley4200测试系统测定实施例1~5以及比较例1~4中制备得到的有机电致 发光器件的驱动电压和电流效率。具体而言,以每秒0.1v的速率提升电压,测定当有机电致发光器件 的亮度达到1000cd/m2时的电压即驱动电压,同时测出此时的电流密度;亮度与电流密度的比值即为 电流效率;
[0178]
实施例1
[0179]
使用本发明化合物a1作为电子传输材料,按照上述有机电致发光器件的制备过程制备有机电致 发光器件,并按照上述有机电致发光器件测试方法进行器件性能测试。
[0180]
实施例2
[0181]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为a2。
[0182]
实施例3
[0183]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为a6。
[0184]
实施例4
[0185]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为a20。
[0186]
实施例5
[0187]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为a38。
[0188]
实施例6
[0189]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为a41。
[0190]
比较例1:
[0191]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为l1。
[0192]
比较例2:
[0193]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为l2。
[0194]
比较例3:
[0195]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为l3。
[0196]
比较例4:
[0197]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为l4。
[0198]
比较例5:
[0199]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为l5。
[0200]
比较例6:
[0201]
采用与实施例1相同的方法制备得到有机电致发光器件,不同在于,将化合物a1替换为l6。
[0202]
本发明上述各个实施例所制备得到的有机电致发光器件的具体性能数据详见下表2:
[0203]
表2:
[0204]
[0205][0206]
就实施例1-6与比较例1-6而言,在有机电致发光器件结构中其他材料相同的情况下,采用本发 明的化合物制备的器件相对于比较例1-6中采用的现有技术中化合物l1-l6所制备的器件的电压均有 所降低,同时采用本发明化合物制备的器件的发光效率相对有较大幅度提升。原因推测可能是本发明 中的代表性化合物含两个缺电子基团,显著提高了电子注入能力。
[0207]
以上实验数据表明,本发明的新型有机材料作为有机电致发光器件的电子传输材料,是性能良好 的有机发光功能材料,有望推广商业化应用。
[0208]
尽管结合实施例对本发明进行了说明,但本发明并不局限于上述实施例,应当理解,在本发明构 思的引导下,本领域技术人员可进行各种修改和改进,所附权利要求概括了本发明的范围。
[0209]
显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域 的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无 法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护 范围之中。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips