
 商标分类
商标分类  商标转让
商标转让 
光致变色材料及其制备方法和应用与流程
 2021-02-02 15:02:41|
2021-02-02 15:02:41| 338|
338| 起点商标网
起点商标网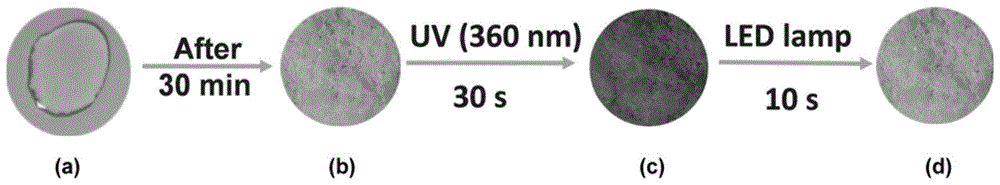
本发明涉及光致变色材料技术领域,尤其是涉及一种光致变色材料及其制备方法和应用。
背景技术:
随着社会的发展假冒伪劣产品给人类社会带来了很大的经济,环境甚至是健康问题。变色材料在一定的紫外激发光刺激下,外观颜色可以快速的发生明显的改变比如从白色迅速变为黄色或者红色,而且在led白光灯的照射下可以快速的回到初始颜色。因而被广泛应用在生物成像,器件、加密防伪等领域。
但是由于传统的光致变色材料会面临材料颜色改变的对比度弱,颜色变化需要的光照时间久,且在几次恢复以后会表现出一定的疲劳性等缺点。最关键的是,由于材料的合成比较复杂导致成本过高以至于很难实现大规模工业化生产。所以很有必要开发出简单,有效且经济的光致变色材料。
有鉴于此,特提出本发明。
技术实现要素:
本发明的第一目的在于提供光致变色材料,以解决现有技术中存在的光致变色材料颜色改变的对比度弱等技术问题。
本发明的第二目的在于提供光致变色材料的制备方法,所述光致变色材料合成工艺简单,条件温和。
本发明的第三目的在于提供光致变色材料的应用,如用于防伪印刷和书写油墨等领域。
为了实现本发明的上述目的,特采用以下技术方案:
光致变色材料,包括化合物a和化合物b;
所述化合物a的结构式如下:
r1选自h、卤素、取代或未取代的含芳基结构;所述含芳基结构包括
r2选自取代或未取代的所述含芳基结构;
所述化合物b的结构式如下:
r3和r4各自独立的选自h、f、cl、br、i、-oh、-och3和-ch3中的任一种。
本发明的光致变色材料,采用化合物a作为客体化合物,化合物b作为主体化合物,将化合物a掺杂到化合物b中,具有明显的光致变色效应。
本发明的光致变色材料在紫外作用下,外观颜色会迅速从白色变为深红色,在黑暗条件下可以维持深红色状态12h以上,在常规的室内光线下可以维持深红色5~10min。并且,在光致变色材料变为深红色后,采用led、白光灯、或手机灯源等照射约10s,光致变色材料从深红色变成白色;且如此往复10次以上,材料仍可保持优异的变色性能。
此外,本发明的化合物a合成简单,化合物b来源广泛,成本低廉,适宜工业化生产。
此外,掺杂材料对湿度、氧气等非常钝感,在潮湿环境下仍然具有非常优异的光致变色效应。
本发明中涉及的*表示基团与化合物其余部分的连接位置。
在本发明的具体实施方式中,取代的含芳基结构中,取代基可以为烷基、烷氧基、卤素基团、酯基中的任一种。如可以为c1~3烷基,包括甲基、乙基、正丙基、异丙基等等。
在本发明的具体实施方式中,所述化合物a的结构式选自如下任一种:
在本发明的具体实施方式中,所述卤素包括f、cl、br和i中的任一种,优选为br。
在本发明的具体实施方式中,所述含芳基结构选自
在本发明的具体实施方式中,所述化合物b为二苯甲酮或4-甲基二苯甲酮。
在本发明的具体实施方式中,所述化合物a的制备方法包括路线a)和路线b)的任一种:
其中,r为r1或r2。
路线a)的具体方法包括:
在室温下,2,6-二甲基吡喃酮与溴化剂如n-溴代琥珀酰亚胺(nbs)进行溴代反应得到化合物ⅰ;
化合物ⅰ与化合物c进行suzuki偶联反应得到化合物ax。
其中,所述suzuki偶联反应的条件包括:化合物ⅰ与化合物c在四三苯基磷钯和碳酸钾的作用下,四氢呋喃和水的混合物作为溶剂,在氮气氛围下于75~85℃反应10~15h。
路线b)的具体方法包括:
在室温下,2,6-二甲基吡喃酮与溴化剂如n-溴代琥珀酰亚胺(nbs)进行溴代反应得到化合物ⅱ;
化合物ⅱ与化合物c进行suzuki偶联反应得到化合物ay1或ay2。
其中,所述suzuki偶联反应的条件包括:化合物ⅱ与化合物c在四三苯基磷钯和碳酸钾的作用下,四氢呋喃和水的混合物作为溶剂,在氮气氛围下于75~85℃反应10~15h。
其中,作为溶剂的四氢呋喃和水的体积比为(5~15)﹕1,如可以为10﹕1。
在实际操作中,在2,6-二甲基吡喃酮nbs的溴代反应中,会同时得到具有单取代溴基团的化合物ⅰ和具有双取代溴基团的化合物ⅱ,采用常规柱层析等分离方式分离得到化合物ⅰ和化合物ⅱ。
在本发明的具体实施方式中,化合物ⅰ与化合物c的摩尔比为1﹕(1~1.5),如可以为1﹕1.2。
在本发明的具体实施方式中,化合物ⅱ与化合物c的摩尔比为1﹕(1~1.5),如可以为1﹕1.2。
在本发明的具体实施方式中,所述四三苯基磷钯的加入量为所述化合物ⅰ的质量的2%~10%,优选为5%;所述碳酸钾的加入量为所述化合物ⅰ的质量的5%~15%,优选为10%。
在本发明的具体实施方式中,所述四三苯基磷钯的加入量为所述化合物ⅱ的质量的2%~10%,优选为5%;所述碳酸钾的加入量为所述化合物ⅱ的质量的5%~15%,优选为10%。
在本发明的具体实施方式中,所述化合物ⅰ与所述溶剂的用量比为1mmol﹕(2~3)ml,如可以为1mmol﹕2.2ml。
在本发明的具体实施方式中,所述化合物ⅱ与所述溶剂的用量比为1mmol﹕(2~3)ml,如可以为1mmol﹕2.2ml。
在实际操作中,在suzuki偶联反应结束后,通过常规柱层析分离提纯的方式得到相应的化合物ax、ay1、ay2。
在本发明的具体实施方式中,所述化合物a和所述化合物b的摩尔比为1﹕(50~20000)。更优选的,所述化合物a和所述化合物b的摩尔比为1﹕(100~2000),更优选为1﹕(200~1000),进一步优选为1﹕1000。
如在不同实施方式中,所述化合物a和所述化合物b的摩尔比可以为1﹕50、1﹕100、1﹕200、1﹕300、1﹕400、1﹕500、1﹕600、1﹕700、1﹕800、1﹕900、1﹕1000、1﹕1200、1﹕1400、1﹕1600、1﹕1800、1﹕2000、1﹕3000、1﹕4000、1﹕5000、1﹕10000、1﹕15000、1﹕20000等等。
本发明还提供了上述光致变色材料的制备方法,包括如下步骤:
所述化合物b于熔融状态下与所述化合物a混合均匀。
本发明采用的化合物b具有较好的可熔融性,在熔融状态下能够溶解化合物a,使化合物a均匀分散在化合物b中。
在本发明的具体实施方式中,所述化合物b于50~60℃的加热条件下熔化至熔融状态。
在本发明的具体实施方式中,还包括:所述混合均匀后,自然冷却至室温。自然冷却到室温使得含有客体分子-化合物a的熔融状态的化合物b缓慢结晶即得到固态的光致变色材料。本发明的制备方法,操作简单,适于大规模生产。
本发明还提供了上述任一种所述光致变色材料在制备油墨和/或墨水中的应用。
进一步的,所述油墨和/墨水为防伪印刷油墨和/或墨水,或者书写绘画油墨和/或墨水。
在实际操作中,可以将上述制备得到的光致变色材料加热至熔融态,直接作为墨水在基材表面进行书法绘画等。其中,所述基材包括打印用纸和滤纸中的任一种。通过该方法得到的书写绘画得到的字样或图案在白光灯下为白色,而在使用紫外光照射后,白色字样或者图案迅速变成深红色,具有高防伪性和加密性。
在本发明的具体实施方式中,所述印刷或书写绘画后,于300~450nm紫外光照射下显色,于500~600nm或者白光照射下恢复颜色。
与现有技术相比,本发明的有益效果为:
(1)本发明的光致变色材料在紫外作用下,外观颜色会迅速从白色变为深红色,在黑暗条件下可以维持深红色状态12h以上,在常规的室内光线下可以维持深红色5~10min。并且,在光致变色材料变为深红色后,采用led、白光灯、或手机灯源等照射约10s,光致变色材料从深红色变成白色;且如此往复10次以上,材料仍可保持优异的变色性能。
(2)本发明的光致变色材料原料来源广泛,合成简单,适于大规模生产。
(3)本发明的光致变色材料可用于防伪印刷和书写油墨,具有较高的防伪性和加密性。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例1提供的光致变色材料的不同光照前后的外观颜色变化示意图;
图2为本发明实施例1提供的光致变色材料在紫外光和led白光灯交替照射处理的紫外吸收重复性;
图3为本发明实施例1提供的光致变色材料在不同温度或水中的不同光照前后的外观颜色变化示意图;
图4为本发明实施例1提供的光致变色材料在不同光照前后的紫外吸收光谱图;
图5为采用本发明实施例1提供的光致变色材料书写的图案在不同光照前后的颜色变化;
图6为本发明实施例2提供的光致变色材料的不同光照前后的外观颜色变化示意图;
图7为本发明实施例2提供的光致变色材料在不同光照前后的紫外吸收光谱图;
图8为采用本发明实施例2提供的光致变色材料书写的图案在不同光照前后的颜色变化;
图9为采用本发明实施例3提供的光致变色材料的不同光照前后的外观颜色变化示意图;
图10为本发明实施例4提供的光致变色材料在不同光照前后的紫外吸收光谱图;
图11为分别采用本发明实施例1提供的光致变色材料和化合物b1书写的图案在不同光照前后的颜色变化;其中“chemis”采用熔融态化合物b1书写,“try”采用熔融态实施例1的光致变色材料书写;
图12为本发明的化合物a2的核磁氢谱图;
图13为本发明的化合物a5的核磁氢谱图;
图14为本发明的化合物a8的核磁氢谱图。
具体实施方式
下面将结合附图和具体实施方式对本发明的技术方案进行清楚、完整地描述,但是本领域技术人员将会理解,下列所描述的实施例是本发明一部分实施例,而不是全部的实施例,仅用于说明本发明,而不应视为限制本发明的范围。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
在本发明的具体实施例中,采用的化合物a的化合物b的结构式如下:
化合物a:
化合物b:
本发明各实施例采用的化合物a的合成路线如下:
包括路线a)和路线b)的任一种:
其中,r为r1或r2。
路线a)的具体方法包括:
在室温下,2,6-二甲基吡喃酮与溴化剂如n-溴代琥珀酰亚胺(nbs)进行溴代反应得到化合物ⅰ;
化合物ⅰ与化合物c进行suzuki偶联反应得到化合物ax。
其中,所述suzuki偶联反应的条件包括:化合物ⅰ与化合物c在四三苯基磷钯和碳酸钾的作用下,四氢呋喃和水的混合物作为溶剂,在氮气氛围下于75~85℃反应10~15h。
路线b)的具体方法包括:
在室温下,2,6-二甲基吡喃酮与溴化剂如n-溴代琥珀酰亚胺(nbs)进行溴代反应得到化合物ⅱ;
化合物ⅱ与化合物c进行suzuki偶联反应得到化合物ay1或ay2。
其中,所述suzuki偶联反应的条件包括:化合物ⅱ与化合物c在四三苯基磷钯和碳酸钾的作用下,四氢呋喃和水的混合物作为溶剂,在氮气氛围下于75~85℃反应10~15h。
其中,作为溶剂的四氢呋喃和水的体积比为(5~15)﹕1,如可以为10﹕1。
在实际操作中,在2,6-二甲基吡喃酮nbs的溴代反应中,会同时得到具有单取代溴基团的化合物ⅰ和具有双取代溴基团的化合物ⅱ,采用常规柱层析等分离方式分离得到化合物ⅰ和化合物ⅱ。
以化合物a1为例,对其合成路线和方法详述如下:
在室温下,2,6-二甲基吡喃酮与n-溴代琥珀酰亚胺(nbs)进行溴代反应得到化合物ⅰ;
然后取10mmol化合物ⅰ与12mmol化合物c1、化合物ⅰ质量的5%的四三苯基磷钯、化合物ⅰ质量的10%的碳酸钾、20ml的四氢呋喃和2ml的水,混合后,于氮气保护氛围下75~85℃条件下反应12h。反应结束后,通过柱层析分离提纯得到目标化合物a1。
化合物a2按照路线b)的第一种进行反应,化合物ⅱ与化合物c1摩尔比为1﹕2.4进行suzuki反应。
化合物a3按照路线b)的第二种进行反应,化合物ⅱ与化合物c1摩尔比为1﹕1.2进行suzuki反应。
化合物a4按照路线a)进行反应,化合物ⅰ与化合物c2摩尔比为1﹕1.2进行suzuki反应;化合物c2的结构式为
化合物a5按照路线b)的第一种进行反应,化合物ⅱ与化合物c2摩尔比为1﹕2.4进行suzuki反应。
化合物a6按照路线b)的第二种进行反应,化合物ⅱ与化合物c2摩尔比为1﹕1.2进行suzuki反应。
化合物a7按照路线a)进行反应,化合物ⅰ与化合物c3摩尔比为1﹕1.2进行suzuki反应;化合物c3的结构式为
化合物a8按照路线b)的第一种进行反应,化合物ⅱ与化合物c3摩尔比为1﹕2.4进行suzuki反应。
化合物a9按照路线b)的第二种进行反应,化合物ⅱ与化合物c3摩尔比为1﹕1.2进行suzuki反应。
化合物a10按照路线a)进行反应,化合物ⅰ与化合物c4摩尔比为1﹕1.2进行suzuki反应;化合物c4的结构式为
化合物a11按照路线b)的第一种进行反应,化合物ⅱ与化合物c4摩尔比为1﹕2.4进行suzuki反应。
化合物a12按照路线b)的第二种进行反应,化合物ⅱ与化合物c4摩尔比为1﹕1.2进行suzuki反应。
化合物a13按照路线a)进行反应,化合物ⅰ与化合物c5摩尔比为1﹕1.2进行suzuki反应;化合物c5的结构式为
化合物a14按照路线b)的第一种进行反应,化合物ⅱ与化合物c5摩尔比为1﹕2.4进行suzuki反应。
化合物a15按照路线b)的第二种进行反应,化合物ⅱ与化合物c5摩尔比为1﹕1.2进行suzuki反应。
通过核磁氢谱和质谱对各化合物的结构进行表征,证实制备得到相应结构的化合物。以化合物a2、a5和a8为例进行说明,其核磁氢谱图分别如图12-图14所示。
实施例1
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a2以及10mmol的化合物b1二苯甲酮,于容器中加热至50℃,使二苯甲酮熔融并将化合物a2溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实施例2
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a5以及10mmol的化合物b1二苯甲酮,于容器中加热至50℃,使二苯甲酮熔融并将化合物a5溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实施例3
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a8以及10mmol的化合物b1二苯甲酮,于容器中加热至50℃,使二苯甲酮熔融并将化合物a5溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实施例4
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a2以及10mmol的化合物b24-甲基二苯甲酮,于容器中加热至60℃,使4-甲基二苯甲酮熔融并将化合物a2溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实施例5
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a1以及10mmol的化合物b1二苯甲酮,于容器中加热至50℃,使二苯甲酮熔融并将化合物a1溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实施例6
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a3以及10mmol的化合物b1二苯甲酮,于容器中加热至50℃,使二苯甲酮熔融并将化合物a3溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实施例7
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a4以及10mmol的化合物b1二苯甲酮,于容器中加热至50℃,使二苯甲酮熔融并将化合物a4溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实施例8
本实施例提供了一种光致变色材料的制备方法,包括如下步骤:
称取0.01mmol的化合物a6以及10mmol的化合物b1二苯甲酮,于容器中加热至50℃,使二苯甲酮熔融并将化合物a6溶解,二者混合均匀后,自然冷却至室温,得到光致变色材料。
实验例1
为了说明本发明的光致变色材料的变色性能,将实施例1的光致变色材料在不同光照前后的外观颜色变化进行检测,如图1所示,其为本发明实施例1提供的光致变色材料的不同光照前后的外观颜色变化示意图。图1(a)为实施例1中将二苯甲酮熔融并将化合物a2溶解(未降至室温)时的照片,其为透明液体;30min后自然冷却至室温,外观如图1(b)所示,形成白色固体;在360nm的紫外光照射30s后,变成红色固体,外观如图1(c)所示;在led白光灯照射10s后,恢复到初始白色固体状态,外观如图1(d)所示。
为了进一步验证本发明的光致变色材料的变色性能的可重复性,取实施例1制备得到的光致变色材料,测试其初始紫外吸收,然后在360nm的紫外光照射30s后,测试其紫外吸收,然后在led白光灯照射10s后,再测试其紫外吸收,交替重复多次。实施例1提供的光致变色材料在紫外光和led白光灯交替照射处理的紫外吸收重复性如图2所示。从图2中可知,本发明的光致变色材料具有较好的可重复性。
实验例2
为了说明本发明的光致变色材料的对湿度等的钝感,将实施例1的光致变色材料在水中测试其在不同光照前后的外观颜色变化,如图3所示,其为本发明实施例1提供的光致变色材料在水中的不同光照前后的外观颜色变化示意图。
以25℃为例进行说明,光致变色材料在25℃条件下,采用360nm紫外光照射30s后,固体颜色由白色变为红色,然后在室内自然光下放置5min,固体颜色保持为红色,然后在led白光灯照射10s后,固体颜色由红色恢复为白色。这是由于室内自然光的强度较弱使得材料在红色状态可以保持一定的时间。
将光致变色材料放入25℃水中浸泡五分钟,过滤得到具有很大湿度的样品。采用360nm紫外光照射30s后,水中粉末的颜色由白色变为红色,然后在自然光下放置5min,固体颜色保持为红色,然后在led白光灯照射10s后,固体颜色由红色恢复为白色。
从上述可知,本发明的光致变色材料对湿度具有钝感。
实验例3
如图4为本发明实施例1提供的光致变色材料在不同光照前后的紫外吸收光谱图。从图中可知,在紫外光照射前与紫外光照射后相比,紫外光照射后,在约530nm处产生新的吸收峰,而在led白光灯照射后,530nm处的新的吸收峰消失。
如图5为采用本发明实施例1提供的光致变色材料书写的图案在不同光照前后的颜色变化。图5(a)为将实施例1的光致变色材料加热至熔融后,直接作为墨水在基材表面进行书写绘画的图案,外观呈白色;在360nm紫外光照射后,外观呈红色,如图5(b)所示;然后再采用led白光灯照射后,外观恢复白色。
实验例4
将实施例2的光致变色材料在不同光照前后的外观颜色变化进行检测,如图6所示,其为本发明实施例2提供的光致变色材料的不同光照前后的外观颜色变化示意图。图6中(a)为实施例2中初始光致变色材料的外观;在360nm紫外光照射30s后,外观呈红色,如图6(b)所示;在暗条件下放置24h,光致变色材料保持红色,外观如图6(c)所示;然后在led白光灯照射10s后,恢复到初始白色固体状态,外观如图6(d)所示。
如图7为本发明实施例2提供的光致变色材料在不同光照前后的紫外吸收光谱图。从图中可知,在紫外光照射前与紫外光照射后相比,紫外光照射后,在约525nm处产生新的吸收峰,而在led白光灯照射后,525nm处的新的吸收峰消失。
如图8为采用本发明实施例2提供的光致变色材料书写的图案在不同光照前后的颜色变化。图8(a)为将实施例2的光致变色材料加热至熔融后,直接作为墨水在基材表面进行书写绘画的图案,外观呈接近白色;在365nm紫外光照射后,外观呈红色,如图8(b)所示;然后再采用led白光灯照射后,外观恢复接近白色。
实验例5
将实施例3的光致变色材料在不同光照前后的外观颜色变化进行检测,如图9所示,其为本发明实施例3提供的光致变色材料的不同光照前后的外观颜色变化示意图。图9中(a)为实施例3中初始光致变色材料的外观;在360nm紫外光照射2min后,外观呈红色,如图9(b)所示;在暗条件下放置12h,光致变色材料保持红色,外观如图9(c)所示;然后在led白光灯照射10s后,恢复到初始白色固体状态,外观如图9(d)所示。
如图10为本发明实施例4提供的光致变色材料在不同光照前后的紫外吸收光谱图。从图中可知,在紫外光照射前与紫外光照射后相比,紫外光照射后,在约525nm处产生新的吸收峰,而在led白光灯照射后,525nm处的新的吸收峰消失。
同理,实施例5-8提供的光致变色材料也具有类似的效果。
实验例6
分别采用本发明实施例1的光致变色材料,化合物b1书写图案,将图案在一定光照条件下,查看实施例1的光致变色材料和化合物b1所书写图案在光照前后的颜色变化情况。具体见图11,图11中的“chemis”采用熔融态化合物b1书写,“try”采用熔融态实施例1的光致变色材料书写。“chemis”在360nm紫外光照射后,外观颜色不变,而“try”在360nm紫外光照射后,图案变为红色。将紫外光照射后的图案,在led白光灯照射后,“chemis”图案颜色仍旧不变,而“try”图案颜色由红色回复为初始颜色。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



