
 商标分类
商标分类  商标转让
商标转让 
一种X射线探测用闪烁体材料及其制备方法与流程
 2021-02-02 14:02:37|
2021-02-02 14:02:37| 234|
234| 起点商标网
起点商标网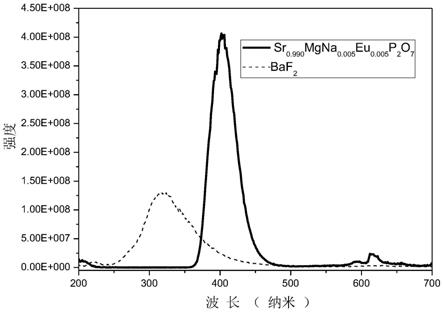
一种x射线探测用闪烁体材料及其制备方法
技术领域
[0001]
本发明涉及x射线探测用闪烁体材料领域,具体涉及一种光产额高、荧光衰减快、不易潮解的一种x射线探测用闪烁体材料及其制备方法。
背景技术:
[0002]
闪烁体材料是一类在x-射线或α,β-射线等高能粒子的照射下可发出紫外或可见光的功能材料,在地质勘探、高能物理和核医学技术方面具有良好的应用前景。性能优良的闪烁体材料应该具有光输出率高、衰减速度快、无余辉等特性。早期用于医学x-ct的闪烁体主要是闪烁单晶,如nai:ti(掺ti的nai单晶),其缺点是存在严重的余辉问题,密度小,易潮解。后来改用znwo4和bgo(bi
4 ge3o
12
),虽然吸收系数大、余辉弱,但光输出小。目前在ct中采用的主要是光输出较大的csi:ti和cdwo4,但csi:ti余辉较长;cd是剧毒元素,且钨酸盐晶体脆性很大,加工时易劈裂。随着x射线层面扫描(x-ct)及正电子发射断层扫描(pet)技术的发展,对闪烁体的性能要求更进一步提高,因此需要不断研制新的闪烁体材料。
[0003]
采用高温固相法合成的碱土金属焦磷酸盐参杂稀土体系srmgnap2o7:eu在空气中能稳定存在,在x-射线激发下具有高的光输出,荧光衰减快。本发明可以为新型闪烁体材料的制备提供理论和技术支持。
技术实现要素:
[0004]
为解决上述问题,本发明旨在公开x射线探测用闪烁体材料领域,具体涉及一种光产额高、荧光衰减快、不易潮解的一种x射线探测用闪烁体材料及其制备方法。
[0005]
为实现上述目的,本发明采用的技术方案是:
[0006]
一种x射线探测用闪烁体材料,其特征在于,所述闪烁体材料的化学组成表示式为:sr
1-2x
eu
x
na
x
mgp2o7,其中eu
2+
为激活离子,x为掺杂离子eu
2+
相对碱土金属离子sr
2+
所占的摩尔百分比系数,取值范围0.001≤x≤0.20。
[0007]
一种x射线探测用闪烁体材料的制备方法,其特征在于,采用高温固相法合成,所述的制备方法包括步骤:
[0008]
1)按照反应配比分别称取原料:碳酸锶、氧化铕、氧化镁、碳酸钠、磷酸氢二铵并混合均匀;
[0009]
2)在研钵中将以上固体混合物充分研磨至无颗粒感,并加入无水乙醇或丙酮将混合粉末进一步混合均匀,反复研磨至颗粒粒径小于5微米;
[0010]
3)在空气气氛中预烧至200~300℃,并在该温度下保持4~6小时,预烧结束后以10~20℃/h的速度降至室温并进一步加入乙醇充分研磨至颗粒粒径小于5微米;
[0011]
4)在空气气氛中以40℃/h的速度升温至700~900℃并保持5~7小时,烧结结束后以40~60℃/h的速度降至室温并进一步加入乙醇充分研磨至颗粒粒径小于5微米后得到产品。
[0012]
优选地,所述步骤1)的原料中,所述磷酸氢二铵可替换为五氧化二磷,或替换为磷
酸氢二铵与五氧化二磷的混合物。
[0013]
优选地,所述步骤1)添加的磷酸氢二铵的含量在原料配比的基础上过量5~25%。
[0014]
优选地,所述步骤2)的所有原料在玛瑙研钵中充分研细并加入乙醇或丙酮混合均匀,反复研磨至原料的颗粒大小为0.1~5微米。
[0015]
优选地,所述步骤3)-4)中,预烧时温度为250℃,升温速度和降温速度均为20℃/h,预烧时间为5小时;而烧结温度为800℃,煅烧时间为6小时,升温速度和降温速度均为40℃/h;预烧和烧结均在空气气氛中进行预烧和烧结,操作更加安全。
[0016]
本发明的有益效果体现在:
[0017]
(1)本发明的闪烁体材料采用高温固相法合成,制备工艺简单,操作安全,条件易于控制;
[0018]
(2)本发明的闪烁体材料,在空气中能稳定存在,不易潮解;
[0019]
(3)本发明所发出的闪烁体材料,荧光波长在360-460nm范围,最强发射峰位于410nm;在x射线激发下,x=0.005时得到的样品光产额为12929ph/mev,为商业baf2晶体的1.33倍。
附图说明
[0020]
图1为sr
0.990
mgna
0.005
eu
0.005
p2o7闪烁发光材料在x射线激发下测得的发射光谱图。
具体实施方式
[0021]
下面结合附图详细说明本发明的具体实施方式:
[0022]
实施例1
[0023]
分别称取碳酸锶(srco3)0.5847g,氧化铕(eu2o3)0.0035g,氧化镁(mgo)0.1612g,碳酸钠(na2co3)0.0011g,磷酸氢二铵((nh4)2hpo4)1.1621g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0024]
实施例2
[0025]
分别称取碳酸锶(srco3)0.5787g,氧化铕(eu2o3)0.0070g,氧化镁(mgo)0.1612g,碳酸钠(na2co3)0.0021g,磷酸氢二铵((nh4)2hpo4)1.1621g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0026]
实施例3
[0027]
分别称取碳酸锶(srco3)0.5669g,氧化铕(eu2o3)0.0141g,氧化镁(mgo)0.1612g,碳酸钠(na2co3)0.0042g,磷酸氢二铵((nh4)2hpo4)1.1621g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入
刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0028]
实施例4
[0029]
分别称取碳酸锶(srco3)0.5551g,氧化铕(eu2o3)0.0211g,氧化镁(mgo)0.1612g,碳酸钠(na2co3)0.0064g,磷酸氢二铵((nh4)2hpo4)1.2677g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0030]
实施例5
[0031]
分别称取碳酸锶(srco3)0.5433g,氧化铕(eu2o3)0.0282g,氧化镁(mgo)0.1612g,碳酸钠(na2co3)0.0085g,磷酸氢二铵((nh4)2hpo4)1.2677g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在一氧化碳还原气氛中于500℃预烧2小时,自然冷却至室温后,将原料倒出在玛瑙研钵中继续充分研磨,再装入刚玉坩埚,在一氧化碳还原气氛中于950℃烧结6小时,自然冷却至室温后研磨均匀,最终得到产品。在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0032]
实施例6
[0033]
分别称取碳酸锶(baco3)0.5315g,氧化铕(eu2o3)0.0352g,氧化镁(mgo)0.1612g,碳酸钠(na2co3)0.0106g,磷酸氢二铵((nh4)2hpo4)1.2677g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0034]
实施例7
[0035]
分别称取碳酸锶(srco3)0.3897g,氧化铕(eu2o3)0.0290g,氧化镁(mgo)0.1209g,碳酸钠(na2co3)0.0087g,磷酸氢二铵((nh4)2hpo4)0.9904g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0036]
实施例8
[0037]
分别称取碳酸锶(srco3)0.3898g,氧化铕(eu2o3)0.0317g,氧化镁(mgo)0.1209g,碳酸钠(na2co3)0.0095g,磷酸氢二铵((nh4)2hpo4)0.9904g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0038]
实施例9
[0039]
分别称取碳酸锶(srco3)0.3809g,氧化铕(eu2o3)0.0370g,氧化镁(mgo)0.1209g,碳酸钠(na2co3)0.0111g,磷酸氢二铵((nh4)2hpo4)0.9904g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0040]
实施例10
[0041]
分别称取碳酸锶(srco3)0.3543g,氧化铕(eu2o3)0.0528g,氧化镁(mgo)0.1209g,碳酸钠(na2co3)0.0159g,磷酸氢二铵((nh4)2hpo4)0.9904g,将上述原料在玛瑙研钵中研磨,并加入无水乙醇作为分散剂,研磨均匀后装入刚玉坩埚,在空气气氛中于250℃预烧4小时,以20℃/h降温至室温后,将原料倒出在玛瑙研钵中继续充分研磨至颗粒小于5微米,再装入刚玉坩埚,在空气气氛中于800℃烧结6小时,以40℃/h降温至室温后研磨均匀至颗粒小于5微米,最终得到产品。
[0042]
上述实施例可以看出,本发明通过控制磷酸氢二铵((nh4)2hpo4)的量(过量10%-20%),在还原气氛中可以获得不同eu
2+
掺杂浓度的sr
1-2x
eu
x
na
x
mgp2o7(x=0.005,0.01,0.02,0.03,0.04,0.05,0.055,0.06,0.07,0.1)闪烁体材料。
[0043]
以上所述,仅是本发明的较佳实施例,并非对本发明的技术范围作任何限制,本行业的技术人员,在本技术方案的启迪下,可以做出一些变形与修改,凡是依据本发明的技术实质对以上的实施例所作的任何修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签: 苏打

 热门咨询
热门咨询




tips