
 商标分类
商标分类  商标转让
商标转让 
一种新型地衣芽孢杆菌宿主细胞及其遗传改造方法与应用与流程
 2021-02-02 13:02:10|
2021-02-02 13:02:10| 322|
322| 起点商标网
起点商标网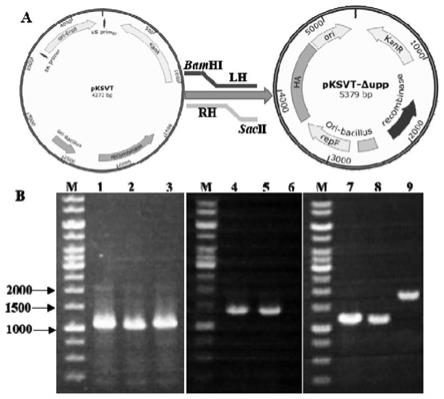
:
[0001]
本发明属于基因工程领域,具体涉及通过对地衣芽孢杆菌 (bacillus licheniformis)2709进行遗传改造获得的新型宿主细胞及其改造方法与应用。
背景技术:
:
[0002]
b.licheniformis 2709工业微生物,来源于土壤等自然环境,进化过程中形成一系列未经驯化的不良特性,包括:1)发酵过程中易形成泡沫,导致极高的发酵污染风险;2)营养缺陷时易形成芽孢,影响目标产物的进一步积累并增加发酵设备的消杀成本;3)形成大量粘性物质,阻碍菌体生长和产物合成,在大规模化工业生产中增加搅拌与溶氧的需求;4)分泌合成大量的胞外蛋白等,这无疑严重影响目标产物的合成与积累,增加了工业化操作的要求与难度,不利于工业化生产的整体效益。
[0003]
近年来,为获得性能优良的工业微生物细胞工厂,学者们在生产菌株的遗传改良方面开展了深入的研究。zhang等人通过敲除影响泡沫形成的脂肽编码基因 srfa,优化了未经驯化的野生菌株b.subtilis atcc 6051a的生产性能;illing等人通过删除芽孢形成关键控制基因sige提高了酶的合成能力;模式菌b.subtiliswb
800
属于多重蛋白酶缺陷型菌株,这明显促进其它胞外分泌蛋白的积累。
[0004]
此外,b.licheniformis理论上可以从基因组精简中获益更多,因为它能够合成大量的对细胞生命活动非必需的次级代谢产物。因此,从分子水平对工业微生物宿主进行分子改良,对微生物细胞生产性能的提升具有重要意义。
[0005]
基因组精简工程主要在原核细胞中进行,尤其是e.coli和b.subtilis等模式菌株,并取得了一定的成果,未见针对工业菌株b.licheniformis 2709基因组精简的报道。分析宿主的基因组发现,存在大量的孤岛基因、冗余基因、假基因等非必需基因,以及控制次级代谢产物合成的基因,这大大增加了细胞代谢负担和能耗,导致营养资源浪费,进而影响细胞的生长与发酵性能。因此,通过小基因组技术实现在特定培养基中底盘细胞的构建,是可行且有价值的。
[0006]
目前,基因组精简工程主要在原核细胞中进行,未见针对工业菌株b. licheniformis 2709基因组精简的报道。分析宿主的基因组发现,存在大量的孤岛基因、冗余基因、假基因等非必需基因,以及控制次级代谢产物合成的基因,这大大增加了细胞代谢负担和能耗,导致营养资源浪费,进而影响细胞的生长与发酵性能。
[0007]
本发明通过分析鉴定影响宿主发酵生产的不良性能,确定相关控制基因,采用建立的遗传操作系统高效删除相关编码基因,以消除宿主细胞不良的生产特征,提升其发酵生产性能;并结合宿主细胞的基因组信息,分析确定可能的孤岛基因等非必需基因,采用小基因组技术,实现有效的基因组精简,为构建适用于工业化生产酶制剂的底盘细胞奠定基础。
技术实现要素:
:
[0008]
为了实现上述目的,本发明将提供一株适用于工业化生产的地衣芽孢杆菌宿主菌,所述菌株是在地衣芽孢杆菌(bacillus licheniformis)2709的基础上,通过敲除eps、pgs、sigf、chiab、pula、amya、lchac、upp基因,并删除9个基因组岛以实现宿主细胞的基因组精简来构建的;
[0009]
所述eps基因位于genbank cp033218中的地衣芽孢杆菌2709即 tccc11148全基因组序列上第1967676-1983603位;
[0010]
所述pgs基因位于genbank cp033218中的地衣芽孢杆菌2709即 tccc11148全基因组序列上第1805920-1808919位;
[0011]
所述sigf基因的genbank编号为cp033218.1(位于全基因组序列上第 3898937-3899704位);
[0012]
所述chiab基因中的chia基因的genbank编号为wp_003178684.1,chib基因的genbank编号为wp_003178682.1(chiab基因位于全基因组序列上第 909808-913767位);
[0013]
所述pula基因的genbank编号为wp_009329394.1(位于全基因组序列上第 2420607-2422739位);
[0014]
所述amya基因的genbank编号为wp_025807921.1(位于全基因组序列上第582978-584516位);
[0015]
所述lchac基因中lcha基因的genbank编号为wp_020450107.1,lchc基因的genbank编号为wp_016886081.1(lchac基因位于全基因组序列上第第834378-849050位);
[0016]
所述upp基因的genbank编号为wp_026589089(位于全基因组序列上第 1714807-1715436位);
[0017]
所述9个基因组岛,分别为位于地衣芽孢杆菌(bacillus licheniformis)2709 基因组上第3,429,766-3,435,147位的gi01;第3,671,385-3,675,741位的gi02;第3,708,534-3,716,479位的gi03;第4,292,644-4,304,905位的gi04;第 4,305,616-4,341,077位的gi05;第1,117,114-1,157,920位的gi06;第 3,560,703-3,567,132位的gi07;第4,071,504-4,088,125位的gi08;第 1,933,894-1,951,991位的gi09,上述位置编号对应genbank cp033218中的地衣芽孢杆菌(bacillus licheniformis)2709即tccc11148全基因组序列位置编码。
[0018]
本发明还提供上述地衣芽孢杆菌宿主细胞的应用,特别是在表达碱性蛋白酶 apre中的应用。
[0019]
有益效果:
[0020]
本发明提供了一种用于b.licheniformis 2709细胞的遗传改造方法,有效改进了该重要工业微生物的发酵生产性能及发酵过程控制。以apre的生产为例, eps、pgs、sigf、chiab、pula、amya、lchac基因,和9个基因组岛敲除菌gr15 对apre的发酵液酶活力可高达32494
±
1013u/ml,高产酶时期长达16h,较对照菌提高了36%。
附图说明:
[0021]
图1 upp基因敲除及验证
[0022]
其中:a,敲除载体的构建流程;b,upp突变体的筛选过程:其中,m是核酸marker,泳
道1-3是载体构建验证结果,1100bp;4-5是单交换验证结果, 1300bp,6是阴性对照,无条带;7-8是双交换验证结果,1250bp,9是阴性对照,1900bp;
[0023]
图2 upp基因敲除流程;
[0024]
图3 upp反向筛选载体构建及验证
[0025]
其中:a,upp反向筛选载体构建过程;b,反向筛选载体的验证:其中,泳道1-3是pcr验证结果,条带大小约为930bp;c是回补重组菌对5-fu的敏感性分析结果;
[0026]
图4 eps突变体的构建
[0027]
其中:a,敲除载体验证:其中,m,marker;泳道1是阴性对照,无条带; 2-4是阳性克隆,条带大小约910bp;b,突变体的筛选验证:其中,泳道1是单交换筛选对照,2-3是正确单交换重组菌,约为1840bp;4-6是正确双交换的突变体,条带大小约为1970bp;
[0028]
图5 chiab缺失对apre表达的影响;
[0029]
图6 chiab,pula和amya缺失对apre表达的影响;
[0030]
图7菌体形态变化;
[0031]
图8不同菌株生长及apre合成情况;
[0032]
图9 blδuppδepsδpgs的生长与产酶分析;
[0033]
图10 blδuppδlch的生长与产酶分析;
[0034]
图11 gr06的产酶能力分析;
[0035]
图12 gr15发酵罐生产碱性蛋白酶。
具体实施方式:
[0036]
为了使本专利的目的、技术方案及优点更加清晰明白,以下结合具体实施例,对本专利进行进一步详细说明。此处所描述的具体实施例仅用以解释本专利,并不用于限定本发明。
[0037]
本发明以穿梭质粒pwh1520及ptu,构建敲除载体;以碱性蛋白酶apre 的表达与分泌情况以及不良发酵性状的有无为指标,敲除了有不良影响或无意义的调控元件,即eps、pgs、sig、chi、pula、amy、lch基因及9个基因组岛对宿主细胞进行基因组精简。相关基因组情况见表1、2。
[0038]
表1宿主性状改良删除的基因信息
[0039][0040]
[0041]
注:upp基因编码尿嘧啶磷酸转移酶,eps基因编码eps合成蛋白酶,pgs基因编码聚谷氨酸合成相关酶,chiab编码几丁质酶合成酶,pula编码普鲁兰酶,amya编码α-淀粉酶, lchac编码地衣素合成酶,sigf编码spo0iiac蛋白。
[0042]
表2本发明分析确定敲除的基因组岛
[0043][0044]
本发明所涉及的引物如表3所示。
[0045]
表3本发明主要引物
[0046]
[0047]
[0048]
[0049]
[0050]
[0051][0052]
note:下划线部分是限制性内切酶
[0053]
本发明涉及的发酵方法如下:
[0054]
发酵培养基:高峰氏淀粉酶0.7g/l,玉米粉64g/l,豆粕40g/l,磷酸氢二钠4g/l,磷酸二氢钾0.3g/l。
[0055]
摇瓶发酵:将菌株在lb平板上进行三区划线,37℃倒置培养过夜,挑取新活化的单菌落于5ml lb培养基中,37℃、220r/min振荡培养12h,以2%的接种量转接于50ml lb液体培养基至od600达0.8-1.0,再以2%的接种量转接于 100ml发酵培养基的挡板瓶中,37℃、220r/min振荡培养,根据实验需要定点取样,4℃、12000r/min离心2min后,取上清液,适当稀释后测定碱性蛋白酶活力。
[0056]
发酵罐发酵:待测菌种接种于装有液体lb培养基(50ml/250ml)的摇瓶中 37℃振荡培养8h。取菌液于脱脂乳板上三区划线,37℃静置培养36h;挑取平板上透明圈相对较大的单克隆至装有液体lb培养基(50/250ml)的摇瓶中,37℃振荡培养8h得种子液,以4%的接种量转接至装有发酵培养基(100/500ml)的摇瓶中,37℃振荡培养7-8h。取培养液以5%的接种量接入3/5l的发酵培养基中,补料控制发酵(糖浓度低于15%时,补料30%糊精,5%棉籽蛋白)。发酵过程中控制ph为7.3,温度为37℃,残糖为15%,溶氧为35%。
[0057]
以下将对过具体实施例对本法做进一步地解释说明。
[0058]
实施例1菌株blδuppδeps的构建
[0059]
1、upp反向筛选载体的构建
[0060]
选择尿嘧啶磷酸核糖转移酶的编码基因upp作为反向筛选标记,需宿主细胞缺失该基因,upp基因敲除载体构建如图1-a所示。利用引物对upplh-f/upplh-r 和upprh-f/upprh-r分别扩增upp基因上、下游同源臂,与bamhi/sacii酶切回收的t2(2)
–
ori质粒片段(质粒及其构建方法公开于中国专利 cn201810898060.8《一种敲除malr的地衣芽孢杆菌菌株及构建方法和应用》中) 重组连接(下文将以pksvt作为t2(2)
–
ori的代称),构成敲除载体pksvt-δupp,采用引物upplh-f/upprh-r进行pcr验证,正确的条带大小约为1100bp(图 1-b),经测序确认后化转至大肠杆菌ec135 pm.bam甲基化修饰,最后电转化至宿主地衣芽孢杆菌2709感受态细胞,25μg/ml kan抗性平板37℃静置培养约 12h。
[0061]
挑取平板上的转化子至相同抗性浓度的5ml的lb培养基中,45℃培养 10h后(1代);吸取10μl菌液转移至同等条件的lb培养基中,45℃继续培养约10h。将培养液稀释涂布kan平板,45℃静置培养,平板上的单克隆即为理论上的单交换菌株。但为避免非特异性整合,采用引物对upp-vf/vr进行菌落pcr验证单交换,结果如图1-b,成功单交换重组菌dna条带大小约为1300 bp,而以感受态基因组为模板的阴性对照无条带,说明发生特异性单交
换。挑取正确单交换的菌落至5ml无抗lb培养基中振荡培养2代约24h(中间转接1 次)。传代培养结束后,将培养液稀释涂布无抗lb平板,37℃静置培养约12h。采用引物upp-vf/upp-vr对平板上单克隆进行菌落pcr验证,如图1-b所示,野生型菌株dna条带大小约为1900bp,而突变株由于upp基因的缺失,扩增的片段大小约为1250bp,说明成功发生双交换,进一步测序验证upp基因删除后的菌株命名为b.licheniformisδupp(以下简称blδupp),作为后续分子改造的出发菌株,据此归纳以温敏型载体和反向筛选标记为基础的基因编辑过程,如图2所示。
[0062]
采用实验室筛选的ps强启动子(ps启动子基因序列: gtcacaatgcgccatcaaaccgttgacaagcgtccccgtcagatggccgggagccggatgaaccaccattccgcgcggc ttgttgacgacaagaacgtcctgatcttattataatataagcaaaaaactcataaaaaggaaaagcattgacctgaaaacttat cggtaaagtatgatataatacaaaaagaccgattagaggggagagaggaaac)来启动自身upp基因的表达,分别以引物对uppps-f/uppps-r和upp-f/upp-r,扩增ps启动子和upp开放阅读框,与经kpni/sali双酶切回收的pksvt载体片段重组连接后,转化至ec135。采用引物uppps-f/upp-r进行pcr验证,结果如图3-b,若构建正确条带大小约为930bp,并进一步测序确认后载体命名为ptu(构建过程见图3-a),即为upp 反向筛选载体。图3-c是回补重组菌对5-fu的敏感性分析结果:将甲基化修饰过的载体ptu和pksvt分别转化blδupp,得到转化子blδupp-u和blδupp-t,分别挑取两种重组菌的单克隆至25μg/ml kan抗性的液体培养基中,37℃振荡培养12h后,检测二者od600,调整为相同od600后分别稀释倾注含 20μg/ml 5-fu、25μg/ml kan和20μg/ml 5-fu+25μg/ml kan三种lb培养基。结果如图3-c,pksvt对blδupp的5-fu敏感性影响较小,重组菌在三种培养基中生长情况差异不显著。ptu的回补则恢复了blδupp对5-fu的敏感性,但当培养基中不加kan时,blδupp-u能够在5-fu平板上部分生长,可能是质粒在无抗性刺激时发生丢失导致的。质粒回补实验表明,upp基因可以作为一种反向筛选标记在blδupp中应用。
[0063]
2、eps基因簇基因敲除
[0064]
利用引物eps-lf/lr以及eps-rf/rr分别扩增eps基因上、下游序列,与酶切回收的载体ptu重组连接,进而得到敲除载体ptu-δeps,载体的酶切验证结果如图4-a所示,酶切验证正确的质粒经甲基化修饰后转化至blδupp。菌落 pcr验证单双交换的结果如图4-b所示,成功单交换的菌株,由于质粒整合到染色体上,利用单交换验证引物eps
–
vf/vr进行pcr扩增,理论上的条带大小约为1840bp;双交换并成功丢失质粒的菌株可以在5-fu平板上生长,采用引物eps-vf/eps-vr进行菌落pcr,成功突变的菌株pcr条带大小约为1970bp,而野生型菌株无条带,pcr结果呈阳性的菌株再次通过pcr和测序,确认正确的敲除菌株命名为blδuppδeps。
[0065]
chiab、pgs、lchac及9个基因组岛敲除方法均同eps基因簇的基因敲除,仅将同源臂引物替换为表3对应引物即可(验证引物也进行对应替换),pula、 amya、sigf敲除为crispr/cas9基因编辑方法,以此来构建表1中各菌株。
[0066]
实施例2不同基因缺失对碱性蛋白酶apre生物合成的影响
[0067]
1、主要胞外蛋白缺失对apre生物合成的影响
[0068]
(1)为考察几丁质酶的缺失对宿主apre分泌表达的影响,分别检测chiab 缺失菌株blδuppδchi和对照菌blδupp发酵培养48h的apre转录水平和apre 酶活力,结果见图
5。结果表明,几丁质酶的缺失对apre的转录水平影响不显著,但其酶活力(13952u/ml)比对照菌(11231u/ml)提高了24.23%。
[0069]
(2)为考察普鲁兰酶(pula)和α-淀粉酶(amya)的缺失对宿主apre 分泌表达的影响,在blδuppδchi的基础上构建pula和amya缺失菌株bl δuppδchiδpulδamy,分别检测blδuppδchiδpulδamy和对照菌blδupp发酵培养48h的apre转录水平和apre酶活力,结果见图6。结果表明,bl δuppδchiδpulδamy的蛋白酶活力(14792u/ml)比对照(11033u/ml)提高34.10%,比blδuppδchi(13952u/ml)提高了5.6%,然而,apre转录水平与对照差异不显著,说明pula和amya的缺失,促进了目标蛋白apre的合成与分泌,进而显著提高发酵液中apre酶活力。
[0070]
2、芽胞缺陷对apre生物合成的影响
[0071]
为研究芽胞起始与形成对blδupp碱性蛋白酶合成的影响,本发明采用 crispr/cas9基因编辑方法(利用单质粒crispr/cas9基因组编辑系统,实现对b.licheniformis 2709基因组的高效编辑。选择sigf作为靶基因,设计敲除载体,相关元件包括:spcas9基因及其启动子p43,特异性靶向sgrna及其转录启动子ps(不含rbs),由分别以sigf-lf、sigf-lr和sigf-rf、sigf-rr为引物合成的500bp左右的上下游同源臂构成的同源修复序列ha,逐一构建至穿梭载体pwh1520上,相关引物及功能见表1-3)),敲除芽胞起始信号控制基因芽胞形成特异性sigma因子编码基因sigf,sigma因子在不对称分裂信号传递后在前胞子内激活表达。缺陷性突变体,命名为blδf。
[0072]
(1)考察突变体在lb液体培养不同时间后菌体形态和芽胞形成率见图7 由图可知,blδf菌体形态变化相似,培养12h时菌体形态与对照菌无显著差异,但培养24h后菌体染色不均匀,有些区域不易着色,但并未形成成熟的芽孢,说明芽胞形成特异性sigma因子的缺失,中断了芽胞成熟的信号。重要的是,芽胞计数结果表明,突变体的芽胞形成率均为0,进一步说明突变体构建成功。
[0073]
(2)为验证不同突变菌株碱性蛋白酶合成能力,我们考察了不同发酵时间 apre的合成情况,检测了不同时间酶活力变化(图8a);同时,为分析导致apre 表达量差异的原因,统计不同产酶时间点的活菌计数,如图8b。结果表明,sigf 缺失突变体的最大酶活力比对照提高24.38%,且在培养40h即达到峰值,可保持产酶稳定期约12h,而对照菌培养46h酶活力才达到峰值,产酶稳定期仅持续约6h。分析菌体生长情况,发现生长稳定期blδf突变体的活菌数高于对照菌,且生长后期生物量下降缓慢。总之,由于突变体因芽胞形成信号的中断而保持良好的蛋白酶合成能力,并使突变体的酶活力下降趋势减慢,且生长后期突变体的生物量明显高于对照菌。这说明,sigf的缺失未影响apre的合成,且使其合成周期大大延长,这有利于工业化生产,并可以提供工业酶制剂生产的优良菌株。
[0074]
3、阻断胞外黏性物质的合成对apre合成的影响
[0075]
为考察普胞外黏多糖(eps)或聚谷氨酸(pga)的缺失对宿主apre分泌表达的影响,构建敲除blδupp中eps基因簇(epsa-epso,基因组位置1967676-1983603,约16kb)的菌株,敲除菌株命名为blδuppδeps;在blδuppδeps的基础上将约3.0kb的γ-pga合成基因簇pgs(基因组位置1805920-1808919)删除。敲除 pgs的突变菌株被命名为blδuppδepsδpgs。
[0076]
检测blδuppδepsδpgs和blδupp发酵培养不同时间的活菌数和碱性蛋白酶活
力变化情况,结果如图9。由图可知,blδuppδepsδpgs生物量比对照菌显著提高18.49%,可能由于聚谷氨酸等粘性物质的消除使发酵液溶氧水平的显著提高,并减少大分子聚合物的合成对atp的消耗,这进而加强了apre的合成,使最大酶活力由11320.7
±
347u/ml提高至14309.3
±
386u/ml,提高了约26.40%;此外,突变菌发酵液溶氧量的增加,使生物量得到有效积累而导致酶活力峰值出现时间(42h)比对照菌提前6h(48h)。发酵液中粘性物质尤其是聚谷氨酸的粘度是高效工业化生产目的蛋白的瓶颈之一,它严重阻碍发酵环境中氧气的传递进而影响胞内可用atp浓度。因此,通过删除控制粘性物质合成的编码基因,可有效阻断粘性物质的分泌合成,避免在生产过程中出现该不良性状而影响目标产物的合成与积累。
[0077]
4、泡沫形成缺陷对apre合成的影响
[0078]
为考察地衣素编码基因lchac的缺失对宿主apre分泌表达的影响,构建敲除载体ptu-δlch,并进一步获得lchac缺失突变菌株blδuppδlch。检测 blδuppδlch和blδupp发酵培养不同时间的活菌数和碱性蛋白酶活力变化情况,结果如图10所示,地衣素缺失突变体和对照菌的生长和蛋白酶合成能力均无显著差异。
[0079]
通过5-l发酵罐分别发酵培养,考察lch缺失对菌株泡沫形成的影响,以及突变体生长和产酶水平的变化。结果表明,突变体的泡沫量大大减少,同等条件下,二者一次性分别需添加消泡剂约30μl和250μl,消泡剂的使用量减少了 87.5%。这足以说明,lch缺失突变体大大减少了泡沫的形成量,有利于发酵过程的控制。
[0080]
5、不良性状编码基因叠加敲除
[0081]
以宿主blδupp为出发菌株,逐一敲除eps、pgs、sigf、chiab、pula、amya、 lchac基因获得菌株blδuppδepsδpgsδsigδchiδpulaδamyδlch,简称gr06。为进一步比较gr06与对照菌blδupp蛋白酶的合成能力,通过摇瓶发酵考察二者合成分泌apre的酶活力,结果如图11,表明gr06的碱性蛋白酶活力(18473
±
503 u/ml)比对照(11225u/ml)提高了约64.57%,且产酶峰值出现时间提前了6-8 h,产酶稳定期比对照延长1倍。因此,影响生产性能的不良性状的整合消除,使优良的发酵性能集合至同一宿主细胞,可大大促进蛋白酶的合成和分泌。
[0082]
实施例3基因组岛的敲除
[0083]
以gr06为出发菌株,采用建立的温敏型基因编辑系统对确定的孤岛基因依次叠加敲除,以t-f/t-r为单交换重组菌验证引物进行敲除验证,如表2所示构建菌株gr07-gr15,并实现基因组精简度达4.3%。为进一步比较gr15与对照菌blδupp蛋白酶的合成能力,通过5l发酵罐发酵考察二者合成分泌apre的酶活力,结果如图12,表明经过5l发酵罐发酵,加强了gr15对apre的合成能力,其酶活力可高达32494
±
1013u/ml,高产酶时期长达16h,而对照菌bl δupp酶活力为23821
±
635u/ml。
[0084]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本专利构思的前提下,上述各实施方式还可以做出若干变形、组合和改进,这些都属于本专利的保护范围。因此,本专利的保护范围应以权利要求为准。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips