
 商标分类
商标分类  商标转让
商标转让 
烷基缩水甘油醚的制备方法与流程
 2021-02-02 13:02:53|
2021-02-02 13:02:53| 438|
438| 起点商标网
起点商标网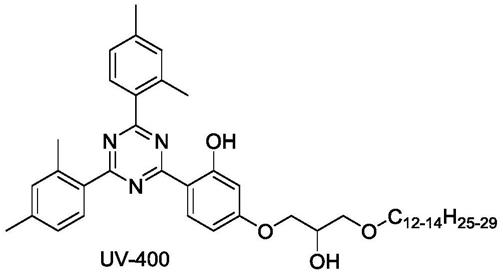
[0001]
本发明涉及有机合成技术领域,具体而言,涉及一种烷基缩水甘油醚的制备方法。
背景技术:
[0002]
uv-400是一种性能优异的液体紫外线吸收剂,主要用于高性能的涂料和汽车漆,其结构如下:
[0003][0004]
uv-400为汽巴公司首先研发成功,其合成所用的两个原料,其一为4,6-二(2,4-二甲苯基)-2-(2,4-二羟基苯基)-1,3,5-三嗪,另一个为混合长碳链烷基缩水甘油醚,即c
12-c
14
烷基缩水甘油醚,uv-400合成路线如下:
[0005][0006]
因此,想要工业化生产紫外线吸收剂uv-400,c
12-c
14
烷基缩水甘油醚的生产技术必须攻克。
[0007]
目前报道的c
12-c
14
烷基缩水甘油醚的合成方法较少,如专利cn101440074中报道了一种c
12-c
14
烷基缩水甘油醚的合成方法,其以两种路易斯酸作为催化剂,三氟化硼乙醚与三氯化铝、四氯化锡和氯化锌中的一种。然而,两种路易斯酸的使用造成催化成本较高,后处理难度较大、三氟化硼毒性大,且最终产品中原料醇的含量约2.5%。而uv400产品对于c
12-c
14
烷基缩水甘油醚的纯度要求大于99%,因此,该缩水甘油醚的合成方法并不适用于c
12-c
14
烷基缩水甘油醚的实际生产。
[0008]
也有一些文献中报道了其他烷基缩水甘油醚的制备方法,比如:刘霜等报道了正新基缩水甘油醚的合成方法(精细与专用化学品,2010,18(9):24-27),该方法以四丁基溴化铵(tbab)为相转移催化剂,甲苯为溶剂,但制得的缩水甘油醚纯度只有87%;专利cn101704730中公开了五种烷基缩水甘油醚的制备方法,该方法以氟硼酸为催化剂,制备的缩水甘油醚收率仅有53~78%;专利cn102971450公开了一种烷基缩水甘油醚的制备方法,此方法采用离子交换膜除去反应过程生成的氯化钠,且需要在工作时向离子交换膜上施加
电流,这导致此方法较难以大规模实施。
[0009]
可见,目前的烷基缩水甘油醚,尤其是混合长碳链烷基缩水甘油醚的制备中普遍存在产品纯度低、收率低等问题。
技术实现要素:
[0010]
本发明的主要目的在于提供一种烷基缩水甘油醚的制备方法,以解决现有技术中制备烷基缩水甘油醚时存在的纯度低、收率低的问题。
[0011]
为了实现上述目的,根据本发明的一个方面,提供了一种烷基缩水甘油醚的制备方法,其特征在于,将烷基醇与环氧卤代丙烷在微通道反应器中进行以下连续流取代反应,得到烷基缩水甘油醚;
[0012][0013]
其中,烷基醇包括式i所示化合物中的一种或多种的混合,且式i中的n为12~14;环氧卤代丙烷具有式ii所示结构,且式ii中的x为卤素原子。
[0014]
进一步地,烷基醇为式i中n为12~14的化合物的混合,环氧卤代丙烷为环氧氯丙烷。
[0015]
进一步地,连续流取代反应在催化剂和液碱的存在下进行;优选地,催化剂选自四丁基溴化铵、乙基三苯基溴化膦和乙基三苯基碘化膦中的一种或多种;优选地,液碱选自氢氧化钠的水溶液。
[0016]
进一步地,制备方法包括以下步骤:将烷基醇与环氧卤代丙烷混合,形成第一原料液;将催化剂与液碱混合,形成第二原料液;将第一原料液与第二原料液送入微通道反应器中以进行连续流取代反应,得到烷基缩水甘油醚。
[0017]
进一步地,连续流取代反应的过程包括:将第一原料与第二原料连续送入微通道反应器中进行取代反应,并将反应得到的取代产物连续排出微通道反应器;提纯取代产物,得到烷基缩水甘油醚。
[0018]
进一步地,连续流取代反应的过程中,第一原料与第二原料的进料速度分别独立地选自10~30ml/min;优选地,第一原料与第二原料的进料速度之比为1:(1~1.5)。
[0019]
进一步地,第一原料液和第二原料液总物料在微通道反应器中的保留时间为1~5min。
[0020]
进一步地,烷基醇与环氧卤代丙烷之间的摩尔比为1:(1~1.5);优选地,液碱中的碱与催化剂之间的摩尔比为1:(0.01~0.05);优选地,烷基醇与催化剂的摩尔比为1:(0.03~0.3)。
[0021]
进一步地,连续流取代反应的温度为50~70℃。
[0022]
进一步地,提纯取代产物的步骤包括:将取代产物静置分层,得到有机相;对有机相进行减压蒸馏,得到烷基缩水甘油醚。
[0023]
本发明提供了一种烷基缩水甘油醚的制备方法,其是将烷基醇与环氧卤代丙烷在微通道反应器中进行以下连续流取代反应,得到烷基缩水甘油醚。采用微通道反应器制备
烷基缩水甘油醚,由于其换热面积大,反应的热效应可控,能够有效提高反应的安全性。同时,在微通道反应器中进行连续流取代反应,反应过程中无返混,反应过程中生成的烷基缩水甘油醚能够及时排除反应器,从而能够有效减少副反应的发生几率,一方面简化了后续的后处理工序,一方面也提高了产品纯度。此外,采用微通道反应器,烷基醇与环氧卤代丙烷能够在反应通道中更充分密切地接触,使得取代反应具有较高的效率和转化率,目标产物的收率也相对较高。
[0024]
总之,本发明有效解决了现有技术中制备烷基缩水甘油醚时反应安全性差、副产物多,以及产品收率和纯度无法兼顾的问题,本发明提供的制备方法操作过程简单、产品纯度高,是一条高效、安全、环保的工艺路线,适合工业化生产。
具体实施方式
[0025]
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将结合实施例来详细说明本发明。
[0026]
正如背景技术部分所描述的,现有技术中制备烷基缩水甘油醚时,纯度低、收率低。
[0027]
为了解决上述问题,本发明提供了一种烷基缩水甘油醚的制备方法,其是将烷基醇与环氧卤代丙烷在微通道反应器中进行以下连续流取代反应,得到烷基缩水甘油醚;
[0028][0029]
其中,烷基醇包括式i所示化合物中的一种或多种的混合,且式ii中的n为12~14;环氧卤代丙烷具有式ii所示结构,且式ii中的x为卤素原子。
[0030]
研究发现,缩水甘油醚的合成过程,伴随着大量的反应热释放,通过反应量热仪测试,混合长碳链烷基缩水甘油醚的合成,反应热可达124kj/mol(以环氧氯丙烷计),绝热温升超过100k。当换热失效后,反应液能达到的温度远超过环氧氯丙烷和液碱的沸点。因此,该产品的合成反应具有非常高的安全风险,对反应器的换热要求极高。此外,由于目标产物中也携带了环氧基团,该基团同样会与原料醇进行反应。因此,采用传统的釜式反应器制备缩水甘油醚,因产物一直在反应釜中进行返混,直至反应结束才能后处理,导致产物中会携带大量的副产物,对后期的提纯也带来较大压力。本发明是将烷基醇与环氧卤代丙烷在微通道反应器中进行以下连续流取代反应,得到烷基缩水甘油醚。采用微通道反应器制备烷基缩水甘油醚,由于其换热面积大,反应的热效应可控,能够有效提高反应的安全性。同时,在微通道反应器中进行连续流取代反应,反应过程中无返混,反应过程中生成的烷基缩水甘油醚能够及时排除反应器,从而能够有效减少副反应的发生几率,一方面简化了后续的后处理工序,一方面也提高了产品纯度(纯度高达99%以上)。此外,采用微通道反应器,烷基醇与环氧卤代丙烷能够在反应通道中更充分密切地接触,使得取代反应具有较高的效率和转化率,目标产物的收率也相对较高。
[0031]
总之,本发明有效解决了现有技术中制备烷基缩水甘油醚时反应安全性差、副产物多,以及产品收率和纯度无法兼顾的问题,本发明提供的制备方法操作过程简单、产品纯
度高,是一条高效、安全、环保的工艺路线,适合工业化生产。
[0032]
此处还需要说明的是,微通道反应器因其反应场所的尺寸特殊性,对于反应体系的性态、原料密度、粘度等方面均具有严格要求,并不是所有的缩水甘油醚都适合采用微通道反应器来进行制备。本发明的烷基醇包括式i所示化合物中的一种或多种的混合,且式ii中的n为12~14;环氧卤代丙烷具有式ii所示结构,且式ii中的x为卤素原子。采用上述烷基醇和环氧卤代丙烷,反应体系更适宜在微通道反应器中进行连续流取代反应。
[0033]
本发明提供的上述制备方法适用于以上烷基缩水甘油醚的制备,当然,考虑到生产过程中放热问题、副产物问题等的严峻程度,更优选将该方法应用于混合长碳链烷基缩水甘油醚的制备,即烷基醇为式i中n为12~14的化合物的混合,环氧卤代丙烷为环氧氯丙烷。混合长碳链烷基缩水甘油醚的制备过程中,对于产品纯度的要求更高,且由于原料中的烷基醇为混合物,对于反应条件的要求更为严苛。而采用微通道反应器进行混合长碳链烷基醇和环氧氯丙烷的连续流取代反应,能够更充分地发挥微通道反应器控热能力强,反应效率高,副产物少等特性,更安全、更绿色、更高效地制备高纯度(大于99%)的混合长碳链烷基缩水甘油醚,能够满足uv 400的使用要求。
[0034]
在一种优选的实施方式中,连续流取代反应在催化剂和液碱的存在下进行。液碱能够及时去除取代反应过程中的小分子酸副产物,再配合催化剂的作用,使得上述取代反应更高效。优选地,催化剂选自四丁基溴化铵、乙基三苯基溴化膦和乙基三苯基碘化膦中的一种或多种;优选地,液碱选自氢氧化钠的水溶液。采用上述几种催化剂和液碱,与烷基醇和环氧氯丙烷形成的反应体系在粘度、密度、流态方面与微通道反应器更相适配,使得连续流取代反应更为稳定。除此以外,上述几种催化剂价格相对较低,有利于降低生产成本。
[0035]
为使连续流取代反应更高效,在一种优选的实施方式中,上述制备方法包括以下步骤:将烷基醇与环氧卤代丙烷混合,形成第一原料液;将催化剂与液碱混合,形成第二原料液;将第一原料液与第二原料液送入微通道反应器中以进行连续流取代反应,得到烷基缩水甘油醚。这样,预先将反应单体混合,将催化气体混合,然后通入微通道反应器中进行反应,有利于各反应原料的充分接触,以便于尽量减少反应时长,在较小的保留时间下获得更高的收率。
[0036]
在实际操作过程中,可以分别将第一原料液和第二原料液置于两个储液单元,然后分别通过驱液装置,比如输送泵,将两个原料液输送至微通道反应器中进行反应。为使反应更稳定,可以在输送管路上各设置一个流量计,以便于监控原料液的输送速度。
[0037]
在一种优选的实施方式中,上述连续流取代反应的过程包括:将第一原料与第二原料连续送入微通道反应器中进行取代反应,并将反应得到的取代产物连续排出微通道反应器;提纯取代产物,得到烷基缩水甘油醚。这样,反应产物及时排出并进行提纯,也减少了副反应的发生几率,更有利于提高产品纯度。具体的提纯过程可以采用本领域的常用类型,但正如前文所述,由于本发明采用了微通道反应器制备烷基缩水甘油醚,反应产物中的副产物较少,可以有效简化提纯工序。在实际操作中,优选上述提纯步骤包括:将取代产物静置分层,得到有机相;对有机相进行减压蒸馏,得到烷基缩水甘油醚。在静置分层之后,也可以采用去离子水对有机相进行洗涤,然后进行减压蒸馏即可。减压蒸馏过程中可以收集180~190℃左右的馏分,即为目标产物烷基缩水甘油醚。
[0038]
为了进一步提高反应效率、产品收率,使烷基醇与环氧卤代丙烷更充分地反应,在
一种优选的实施方式中,连续流取代反应的过程中,第一原料与第二原料的进料速度分别独立地选自10~30ml/min;优选地,第一原料与第二原料的进料速度之比为1:(1~1.5)。
[0039]
在一种优选的实施方式中,第一原料液和第二原料液总物料在微通道反应器中的保留时间为1~5min。将保留时间控制在上述范围内,更有利于兼顾反应效率、产品纯度等方面。优选地,连续流取代反应的温度为50~70℃。在实际操作过程中,可以预先将微通道反应器升温至反应温度,其次再将原料液通入其中进行反应。比如,可以通过换热系统将热油通入微通道反应器的控温夹套,待热油出口温度稳定后,再打开原料液的输送管路,具体操作时本领域技术人员可以理解的,在此不再赘述。
[0040]
在一种优选的实施方式中,烷基醇与环氧卤代丙烷之间的摩尔比为1:(1~1.5);优选地,液碱中的碱与催化剂之间的摩尔比为1:(0.01~0.05);优选地,烷基醇与催化剂的摩尔比为1:(0.03~0.3)。将各原料的用量关系控制在上述范围内,一方面更有利于提高取代反应的效率,另一方面原料液在进入微通道反应器之后,整个反应体系更为稳定。
[0041]
以下结合具体实施例对本申请作进一步详细描述,这些实施例不能理解为限制本申请所要求保护的范围。
[0042]
实施例1
[0043]
该实施例中制备了混合长碳链烷基缩水甘油醚,具体操作如下:
[0044]
于常温(约25℃)下,将196g(约1.0mol)c
12-c
14
长链混合烷基醇(商购)、111g(1.2mol)环氧氯丙烷加入到单口烧瓶中,磁力搅拌,至物料完全混合,形成第一原料液;将48g(1.2mol)naoh、300ml水和11g(0.03mol)乙基三苯基溴化膦加入到单口圆底烧瓶中,电磁搅拌,至完全混合,形成第二原料液;
[0045]
打开微通道反应器的换热系统,设定热油温度为50℃,至微通道反应器的热油出口温度为50
±
0.5℃后开动两个加料泵a和b,加料泵a输送第一原料液,加料泵b输送第二原料液。其中,第一原料液和第二原料液的输料速度均为20ml/min,经过6块微通道反应模块(每块模块的容积为8ml,顺次串联,且孔道折叠排列),第一原料液和第二原料液总物料在微通道反应器中的保留时间为3.2min,进行连续流取代反应,反应过程中连续进料,连续出料。
[0046]
用1000ml四口圆底烧瓶接收反应液,后反应液静置,分出下层水相,并加入少量水,水洗一次,有机相进行减压蒸馏,收集180-190℃馏分(2-3mmhg),产物为无色、略粘稠的液体,收率85%,气相色谱含量99.4%。
[0047]
产物gcms:c
15
h
30
o
2
[m+]=242.40(c12烷基缩水甘油醚),c
17
h
34
o
2
[m+]=270.46(c14烷基缩水甘油醚),离子峰,111,125,139,153,171,185,227。红外光谱波数(cm-1):2955,2919,2865,1466,1384,1248,1103,912,839,758。
[0048]
实施例2
[0049]
与实施例1不同之处在于:
[0050]
打开微通道反应器的换热系统,设定热油温度为60℃至微通道反应器的热油出口温度为60
±
0.5℃后开动两个加料泵a和b,加料泵a输送第一原料液,加料泵b输送第二原料液。其中,第一原料液和第二原料液的输料速度均为20ml/min,物料的保留时间1.2min,连续进料,连续出料。
[0051]
用1000ml四口圆底烧瓶接收反应液,后反应液静置,分出下层水相,并加入少量
水,水洗一次,有机相进行减压蒸馏,收集180-190℃馏分(2-3mmhg),产物为无色、略粘稠的液体,收率83%,气相色谱含量99.6%。
[0052]
实施例3
[0053]
与实施例1不同之处在于:
[0054]
打开微通道反应器的换热系统,设定热油温度为60℃至微通道反应器的热油出口温度为60
±
0.5℃后开动两个加料泵a和b,加料泵a输送第一原料液,加料泵b输送第二原料液。其中,第一原料液和第二原料液的输料速度均为15ml/min,物料的保留时间1.6min,连续进料,连续出料。
[0055]
用1000ml四口圆底烧瓶接收反应液,后反应液静置,分出下层水相,并加入少量水,水洗一次,有机相进行减压蒸馏,收集180-190℃馏分(2-3mmhg),产物为无色、略粘稠的液体,收率86.6%,气相色谱含量99.7%。
[0056]
实施例4
[0057]
与实施例1不同之处在于:
[0058]
于常温(约25℃)下,将196g(约1.0mol)c
12-c
14
长链混合烷基醇、139g(1.5mol)环氧氯丙烷加入到单口烧瓶中,磁力搅拌,至物料完全混合,形成第一原料液;将48g(1.2mol)naoh、300ml水和9.67g(0.03mol)四丁基溴化铵,加入到单口圆底烧瓶中,电磁搅拌,至完全混合,形成第二原料液;
[0059]
打开微通道反应器的换热系统,设定热油温度为60℃至微通道反应器的热油出口温度为60
±
0.5℃后开动两个加料泵a和b,加料泵a输送第一原料液,加料泵b输送第二原料液。其中,第一原料液和第二原料液的输料速度均为15ml/min,物料的保留时间1.6min,连续进料,连续出料。
[0060]
用1000ml四口圆底烧瓶接收反应液,后反应液静置,分出下层水相,并加入少量水,水洗一次,有机相进行减压蒸馏,收集180-190℃馏分(2-3mmhg),产物为无色、略粘稠的液体,收率87.4%,气相色谱含量99.5%。
[0061]
实施例5
[0062]
与实施例1不同之处在于:
[0063]
于常温(约25℃)下,将196g(约1.0mol)c
12-c
14
长链混合烷基醇(商购)、93g(1mol)环氧氯丙烷加入到单口烧瓶中,磁力搅拌,至物料完全混合,形成第一原料液;将240g(6mol)naoh、300ml水和110g(0.3mol)乙基三苯基溴化膦加入到单口圆底烧瓶中,电磁搅拌,至完全混合,形成第二原料液;
[0064]
打开微通道反应器的换热系统,设定热油温度为60℃至微通道反应器的热油出口温度为60
±
0.5℃后开动两个加料泵a和b,加料泵a输送第一原料液,加料泵b输送第二原料液。其中,第一原料液和第二原料液的输料速度均为30ml/min,物料的保留时间1.6min,连续进料,连续出料。
[0065]
用1000ml四口圆底烧瓶接收反应液,后反应液静置,分出下层水相,并加入少量水,水洗一次,有机相进行减压蒸馏,收集180-190℃馏分(2-3mmhg),产物为无色、略粘稠的液体,收率85.6%,气相色谱含量99.2%。
[0066]
实施例6
[0067]
与实施例1不同之处在于:
[0068]
于常温(约25℃)下,将196g(约1.0mol)c
12-c
14
长链混合烷基醇(商购)、111g(1.2mol)环氧氯丙烷加入到单口烧瓶中,磁力搅拌,至物料完全混合,形成第一原料液;将120g(3mol)naoh、300ml水和11g(0.03mol)乙基三苯基溴化膦加入到单口圆底烧瓶中,电磁搅拌,至完全混合,形成第二原料液;
[0069]
打开微通道反应器的换热系统,设定热油温度为60℃至微通道反应器的热油出口温度为60
±
0.5℃后开动两个加料泵a和b,加料泵a输送第一原料液,加料泵b输送第二原料液。其中,第一原料液和第二原料液的输料速度均为30ml/min,物料的保留时间1.6min,连续进料,连续出料。
[0070]
用1000ml四口圆底烧瓶接收反应液,后反应液静置,分出下层水相,并加入少量水,水洗一次,有机相进行减压蒸馏,收集180-190℃馏分(2-3mmhg),产物为无色、略粘稠的液体,收率83.5%,气相色谱含量99.3%。
[0071]
对比例1
[0072]
于常温(约25℃)下,将196g(约1.0mol)c
12-c
14
长链混合烷基醇、9.67g(0.03mol)四丁基溴化铵、48g(1.2mol)naoh和300ml水加入到1000ml四口烧瓶中,滴加139g(1.5mol)环氧氯丙烷,用时2h,滴加完后40℃保温1h,后升温至60℃,保温反应3h,停止反应。反应结束后分液,分掉下层水相,有机相再用水洗3次,至有机相呈中性,后减压蒸馏,收集180-190℃馏分(2-3mmhg),产物为无色、略粘稠的液体,收率67.4%,气相色谱含量99.4%。
[0073]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips