
 商标分类
商标分类  商标转让
商标转让 
一种基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法与流程
 2021-02-02 13:02:42|
2021-02-02 13:02:42| 365|
365| 起点商标网
起点商标网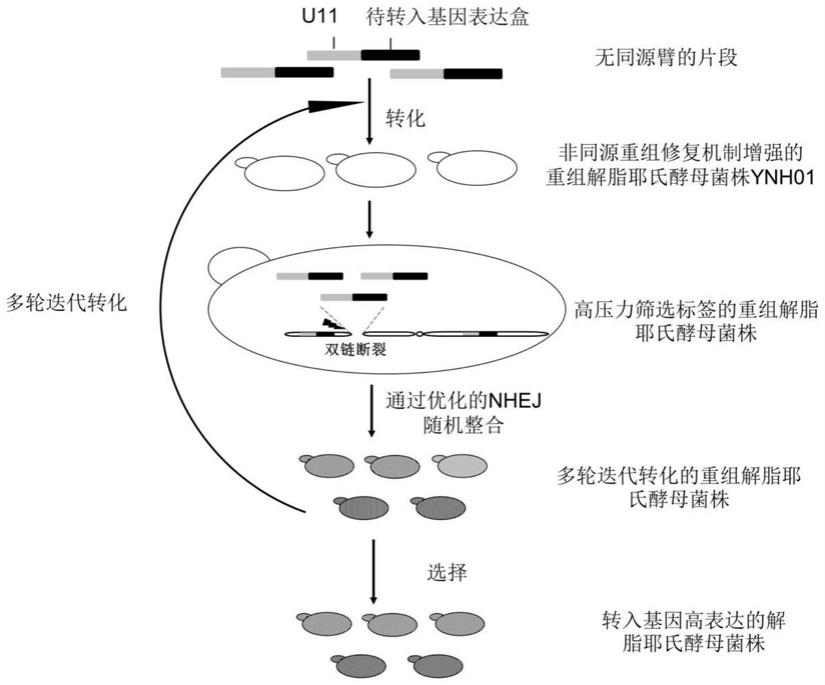
[0001]
本发明涉及生物技术领域,更具体的说是涉及一种基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法。
背景技术:
[0002]
在生物体中,dna分子作为生命信息的载体,在遗传信息的存储、复制及传递过程中有着至关重要的作用。dna分子的损伤严重影响细胞的生长及功能,其中,dna双链断裂(dna double-strand breaks,dsbs)是最有害的dna损伤之一,不仅会导致潜在的序列信息丢失,还会影响细胞的生长状态。为了有效修复生长过程中发生的dna双链断裂,细胞主要通过两种途径实现,分别为同源重组(homologous recombination,hr)和非同源末端连接(non-homologous end-joining,nhej)。dsb两种修复途径的根本区别在于是否需要同源dna序列。同源重组过程需要一段同源序列,在一系列关键酶的催化作用下,将断裂的dna分子双链重接整合起来,这种修复方式在原核生物及低等真核生物的dsbs修复中起主要作用。而在高等真核生物中,dsbs主要通过非同源末端连接的方式进行修复。与同源重组不同的是,非同源重组无需任何序列同源性,直接将无同源性的dna末端连接起来。尽管已经在一些真核生物(如酿酒酵母)中发现了nhej机制,但在这些生物中它仅用作修复dsbs的备用系统,占主导作用的仍是同源重组机制。
[0003]
近年来,解脂耶氏酵母逐渐引起研究人员的关注,不仅拥有强大的脂质合成能力及异源蛋白表达能力,还能够利用廉价底物合成价值较高的化合物,逐渐成为具有潜力的底盘菌株被广泛应用于细胞工厂的生产中。由于解脂耶氏酵母的同源重组效率远低于酿酒酵母,非同源重组效率占主要地位,这些常规的生物技术在解脂耶氏酵母中无法实现有效的应用。因此,探索优化解脂耶氏酵母的非同源末端连接机制,在解脂耶氏酵母中建立nhej整合技术,是亟需解决的问题。
技术实现要素:
[0004]
本发明的目的在于克服现有技术的不足,提供一种基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法。
[0005]
本发明的技术方案概述如下:
[0006]
一种基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法,包括如下步骤:
[0007]
(1)将解脂耶氏酵母内源基因dl4和xrcc4,外源智人基因paxx以rdna位点整合的方式整合到解脂耶氏酵母atcc201249菌株的基因组rdna位点,得到解脂耶氏酵母非同源重组修复机制增强的重组菌株ynh01;
[0008]
所述dl4基因的核苷酸序列如seq id no.1所示;
[0009]
所述paxx基因的核苷酸序列如seq id no.2所示;
[0010]
所述xrcc4基因的核苷酸序列如seq id no.3所示;
[0011]
(2)将待转入基因表达盒与启动子截短到11bp的ura3营养标签u11相连接,得到连接片段;将所述连接片段转化到所述重组菌株ynh01,得到高压力筛选标签菌株;
[0012]
所述启动子截短到11bp的ura3营养标签u11核苷酸序列如seq id no.49所示;
[0013]
(3)将所述连接片段再次转化到高压力筛选标签菌株,得到经过两轮转化的重组菌株;
[0014]
(4)将所述连接片段再次转化到经过两轮转化的重组菌株,得到转入的基因高表达的菌株。
[0015]
待转入基因表达盒为绿色荧光蛋白gfp基因表达盒或番茄红素合成基因crte、crtb和crti表达盒;
[0016]
所述绿色荧光蛋白gfp基因表达盒的核苷酸序列如seq id no.4所示;
[0017]
所述crte基因的核苷酸序列如seq id no.5所示;
[0018]
所述crtb基因的核苷酸序列如seq id no.6所示
[0019]
所述crti基因的核苷酸序列如seq id no.7所示。
[0020]
本发明的优点:
[0021]
本发明一种基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法,在短时间内获得大量的多样性菌株。利用本发明的方法,建立以番茄红素的异源合成为代表的途径,成功得到一系列产量差异显著的菌株。实现了nhej整合技术在天然产物微生物合成中的高效应用。
附图说明
[0022]
图1为基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法概念图。
[0023]
图2为优化关键连接酶非同源重组修复机制增强的重组菌株ynh01绿色荧光蛋白荧光强度图。
[0024]
图3为优化高压力筛选标签的重组解脂耶氏酵母菌株绿色荧光蛋白荧光强度图。
[0025]
图4为优化多轮迭代转化的重组解脂耶氏酵母菌株绿色荧光蛋白荧光强度图。
[0026]
图5为利用本发明的方法合成番茄红素重组解脂耶氏酵母菌株番茄红素产量图。
具体实施方式
[0027]
原始菌株解脂耶氏酵母yarrowia lipolytica atcc201249,即解脂耶氏酵母菌株yarrowia lipolytica(wickerham et al.)van der walt et von arx(201249
tm
)于2014年9月在atcc官网购买。
[0028]
(https://www.atcc.org/products/all/201249.aspx)
[0029]
向解脂耶氏酵母atcc201249菌株中转入绿色荧光蛋白gfp基因表达盒(seq id no.4)得到对照菌株ynh00。
[0030]
dl4基因:来自解脂耶氏酵母(yarrowia lipolytica clib122)
[0031]
paxx基因:来自智人(homo sapiens)
[0032]
xrcc4基因:来自解脂耶氏酵母(yarrowia lipolytica clib122)
[0033]
gfp基因:来自合成型构建(synthetic construct)
[0034]
crte基因:来自斯塔瓦特氏欧文氏菌(pantoea stewartii dc413)
[0035]
crtb基因:来自斯塔瓦特氏欧文氏菌(pantoea stewartii dc413)
[0036]
crti基因:来自斯塔瓦特氏欧文氏菌(pantoea stewartii dc413)
[0037]
实施例1
[0038]
一种基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法(见图1),包括如下步骤:
[0039]
(1)从ncbi数据库获得yarrowia lipolytica clib122来源的基因dl4(seq id no.1)和xrcc4(seq id no.3),homo sapiens来源的基因paxx氨基酸序列,通过密码子优化人工合成paxx基因(seq id no.2)的序列,同时组装三个基因的表达盒。
[0040]
通过同源重组的方法,向解脂耶氏酵母atcc201249菌株的基因组rdna位点整合了dl4基因表达盒,paxx基因表达盒和xrcc4基因表达盒,得到解脂耶氏酵母非同源重组修复机制增强的重组菌株ynh01。
[0041]
以ynh01为底盘转入绿色荧光蛋白gfp基因表达盒(seq id no.4),观察荧光强度(见图2)。
[0042]
(2)第一种高压力筛选标签菌株的构建方法,包括如下步骤:
[0043]
将ura3标签进行扩增,上游引物的结合位点设置在ura3启动子的不同位置处,分别在启动子的41bp、21bp、16bp、15bp、14bp、11bp、9bp、8bp及6bp处,得到一系列突变标签,命名为u41、u21、u16、u15、u14、u11、u9、u8及u6。
[0044]
将截短启动子之后的ura3标签与绿色荧光蛋白gfp基因表达盒(seq id no.4)进行连接,得到相应的一系列重组片段,将上述9个线性片段分别转入重组菌株ynh01中测量荧光强度。其中绿色荧光蛋白gfp基因表达盒(seq id no.4)与启动子截短到11bp的ura3营养标签u11(seq id no.49)相连接得到的片段命名为连接片段-1,将连接片段-1转化到重组菌株ynh01后得到最优化第一种高压力筛选标签菌株(见图3)。
[0045]
(3)第一种多轮迭代转化重组解脂耶氏酵母菌株的构建,包括如下步骤:
[0046]
将上一步得到的最优化第一种高压力筛选标签菌株的300μl的转化体系接入50ml液体ypd培养基的250ml锥形瓶中,于28℃、250rpm的条件下培养36h。待摇瓶中的菌株生长至od600为0.6-0.8时,用无菌水洗涤2次,于4500rpm/min离心1分钟,弃去上清后,用连接片段-1进行第二轮转化,得到第一种经过两轮转化的重组菌株,以上述操作流程为准,将连接片段-1再次转化上一步得到的第一种经过两轮转化的重组菌株最终得到经过三轮转化后绿色荧光蛋白gfp基因高表达的菌株。
[0047]
在第二轮转化结束后,接入50ml液体ypd培养基中的体系只需培养24h。主要是由于前两轮的迭代转化,使得菌株数量有所积累,od600达到0.6-0.8所需时间会缩短。同理,第3轮转化结束后,接入50ml液体ypd中的体系,只需12h即可进行第4轮转化。经过多轮迭代转化后,挑取每一轮固体培养基上的转化子,接入无菌96孔板上进行培养发酵,48h后测定荧光值(见图4)。
[0048]
实施例2
[0049]
一种基于非同源末端连接机制的解脂耶氏酵母基因组整合的方法,包括如下步骤:
[0050]
(1)同实施例1步骤(1)重组菌株ynh01的制备;
[0051]
(2)第二种高压力筛选标签菌株的构建方法,包括如下步骤:
[0052]
将启动子截短到11bp的ura3营养标签u11与番茄红素合成基因crte(seq id no.5)表达盒h0-ld01-crte-h1、番茄红素合成基因crtb(seq id no.6)表达盒h1-ld02in-crtb-h2和番茄红素合成基因crti(seq id no.7)表达盒h2-ld03-crti-h3,4个片段相连接得到连接片段-2;将连接片段-2转化到重组菌株ynh01后,得到第二种高压力筛选标签菌株;
[0053]
crte(seq id no.5)、crtb(seq id no.6)和crti(seq id no.7)是来自斯塔瓦特氏欧文氏菌pantoea stewartii dc413经基因密码子优化后人工合成的;
[0054]
(3)第二种多轮迭代转化重组解脂耶氏酵母菌株的构建,包括如下步骤:
[0055]
将上一步得到的最优化第二种高压力筛选标签菌株的300μl的转化体系接入50ml液体ypd培养基的250ml锥形瓶中,于28℃、250rpm的条件下培养36h。待摇瓶中的菌株生长至od600为0.6-0.8时,用无菌水洗涤2次,于4500rpm/min离心1分钟,弃去上清后,用连接片段-2进行第二轮转化,得到第二种经过两轮转化的重组菌株,以上述操作流程为准,将连接片段-2再次转化上一步得到的经过第二种两轮转化的重组菌株最终得到经过三轮转化后番茄红素合成基因高表达的菌株。
[0056]
将启动子截短到11bp的ura3营养标签u11、h0-ld01-crte-h1;h1-ld02in-crtb-h2;h2-ld03-crti-h3四个片段转化到出发菌株解脂耶氏酵母atcc201249中,得到对照菌株nhej-ly00。
[0057]
另一方面,为了与同源重组整合效率进行对比,我们将带有rdna位点同源臂的
[0058]
rdnaup-hyg-h0(rdna位点上同源臂片段);
[0059]
h0-ld01-crte-h1;
[0060]
h1-ld02in-crtb-h2;
[0061]
h2-ld03-crti-h3;
[0062]
h3-rdna-down(rdna位点下同源臂片段)
[0063]
5个片段转化到出发菌株解脂耶氏酵母atcc201249中,经过菌落pcr及测序验证正确整合后,得到成功通过同源重组方式整合到rdna位点的转化子hr-ly00系列对照菌株,菌株接入50ml的ypd发酵培养基的250ml锥形瓶中进行发酵,72h后提取番茄红素,用高效液相色谱测定番茄红素的产量(见图5)。
[0064]
1、模块的构建
[0065]
同源臂rdna-up(seq id no.14)和rdna-down(seq id no.15)均来自解脂耶氏酵母atcc201249基因组;
[0066]
筛选标记基因ura3(seq id no.8)和潮霉素b抗性基因hyg表达盒(seq id no.9)来自天津大学元英进教授元件库。
[0067]
以解脂耶氏酵母atcc 201249基因组为模板,以rdnaup-f(seq id no.10),rdnaup-hyg-r(seq id no.11)以及rdnadown-h3-f(seq id no.12),rdnadown-r(seq id no.13)为引物,分别扩增同源臂得到rdna-up(seq id no.14)和rdna-down(seq id no.15)
[0068]
以h3-f(seq id no.16)和h3-r(seq id no.17)分别为前后引物,yl-ld03载体(seq id no.21)为模板,扩增rdna位点基因组下臂和yl-ld03载体(seq id no.21)之间连接的同源臂h3(seq id no.18);
[0069]
将rdna-up(seq id no.14)和潮霉素b抗性基因hyg表达盒(seq id no.9)通过overlap pcr连接得到rdnaup-hyg-h0线性片段;
[0070]
将rdna-down(seq id no.15)和h3(seq id no.18)通过overlap pcr连接得到h3-rdna-down线性片段;
[0071]
将yl-ld01(seq id no.19)载体用bsai酶切后与dl4(seq id no.1)基因连接,然后再用noti酶切释放得到h0-ld01-dl4-h1线性片段;
[0072]
将yl-ld02in(seq id no.20)载体用bsmbi酶切后与paxx(seq id no.2)基因连接,然后再用noti酶切释放得到h1-ld02in-paxx-h2线性片段;
[0073]
将yl-ld03(seq id no.21)载体用bsai酶切后与xrcc4(seq id no.3)基因连接,然后再用noti酶切释放得到h2-ld03-xrcc4-h3线性片段;
[0074]
然后将这些片段按照每个片段的摩尔比为1:1,片段总量达到1μg,使用frozen-ez yeast transformation ii试剂盒(zymo research公司)转化解脂耶氏酵母。在添加潮霉素b(hyg终浓度100mg/l)的ypd固体培养基(20g/l葡萄糖,20g/l蛋白胨,10g/l酵母提取物,20g/l琼脂)涂板48h后挑选正确的转化子,构建得到重组菌株ynh01。
[0075]
gfp绿色荧光蛋白基因表达盒(seq id no.4)和完整ura3(seq id no.8)筛选标签通过overlap pcr连接得到gfp-ura3线性片段;所用的两对引物分别为(gfp-f(seq id no.22);gfp-ura-r(seq id no.23);ura-gfp-f(seq id no.24);ura-r(seq id no.25));将1μg的gfp-ura3线性片段使用frozen-ez yeast transformation ii试剂盒(zymo research公司)转化解脂耶氏酵母菌株ynh01,涂板sc-ura固体培养基(20g/l葡萄糖,2g/l不含尿嘧啶的氨基酸混合物,6.7g/l不含氨基酸的酵母氮碱基,20g/l琼脂),在28℃下孵育36h后,挑单菌落在28℃,250rpm的ypd液体培养基(20g/l葡萄糖,20g/l蛋白胨,10g/l酵母提取物,20g/l琼脂固体培养基)中培养过夜。
[0076]
用前引物gfp-f(seq id no.22)分别和后引物gfp
–
u41-r(seq id no.26);gfp-u21-r(seq id no.27);gfp-u16-r(seq id no.28);gfp-u15-r(seq id no.29);gfp-u14-r(seq id no.30);gfp-u11-r(seq id no.31);gfp-u9-r(seq id no.32);gfp-u8-r(seq id no.33);gfp-u6-r(seq id no.34)扩增gfp表达盒片段用于和各种突变的ura3标签连接;
[0077]
再用前引物u41-gfp-f(seq id no.35);u21-gfp-f(seq id no.36);u16-gfp-f(seq id no.37);u15-gfp-f(seq id no.38);u14-gfp-f(seq id no.39);u11-gfp-f(seq id no.40);u9-gfp-f(seq id no.41);u8-gfp-f(seq id no.42);u6-gfp-f(seq id no.43)分别和后引物ura-r(seq id no.25)扩增突变ura3标签得到突变标签u41;u21;u16;u15;u14;u11;u9;u8和u6。将上一步扩增的gfp表达盒片段和突变标签u41;u21;u16;u15;u14;u11;u9;u8;u6分别连接得到对应的线性片段gfp-u41、gfp-u21、gfp-u16、gfp-u15、gfp-u14、gfp-u11、gfp-u9、gfp-u8及gfp-u6。将各个线性片段转化解脂耶氏酵母ynh01,测量荧光强度。
[0078]
将yl-ld01(seq id no.19)载体用bsai酶切后与crte(seq id no.5)基因连接,然后再用noti酶切释放得到h0-ld01-crte-h1线性片段;
[0079]
将yl-ld02in(seq id no.20)载体用bsmbi酶切后与crtb(seq id no.6)基因连接,然后再用noti酶切释放得到h1-ld02in-crtb-h2线性片段;
[0080]
将yl-ld03(seq id no.21)载体用bsai酶切后与crti(seq id no.7)基因连接,然
后再用noti酶切释放得到h2-ld03-crti-h3线性片段;
[0081]
以h0-u11-f(seq id no.44)和h0-r(seq id no.45)分别为前后引物扩增筛选标记基因ura3(seq id no.8)和yl-ld01载体之间连接的同源臂h0(seq id no.46);
[0082]
以ura3(seq id no.8)为模板,分别以u11-h0-f(seq id no.47)、u11-h0-r(seq id no.48)为引物扩增标记基因u11(seq id no.49),通过overlap pcr将片段u11(seq id no.49)和h0(seq id no.46)连接得到线性片段u11-h0;
[0083]
然后将u11-h0;h0-ld01-crte-h1;h1-ld02in-crtb-h2;h2-ld03-crti-h3这4个片段按照每个片段的摩尔比为1:1,片段总量达到1μg,使用frozen-ez yeast transformation ii试剂盒(zymo research公司)转化解脂耶氏酵母菌株ynh01为实验组;转化解脂耶氏酵母菌株ynh00为nhej对照组。将片段rdnaup-hyg-h0;h0-ld01-crte-h1;h1-ld02in-crtb-h2;h2-ld03-crti-h3;h3-rdna-down这5个片段按照每个片段的摩尔比为1:1,片段总量达到1μg转化解脂耶氏酵母菌株atcc201249作为为通过同源重组整合这些片段的对照组。
[0084]
所用序列见序列表。
[0085]
本发明所用pcr酶为南京诺唯赞生物科技有限公司的super-fidelity聚合酶。50μl的pcr扩增体系如下:dna模板,1μl;前引(10μm)和后引(10μm)各2μl;dntp(10mm),1μl;2
×
phanta max buffer,20μl;super-fidelity聚合酶,1μl;最后用双蒸水补齐至50μl。在pcr仪上设置扩增程序。扩增条件为95℃预变性3min(1个循环);95℃变性15sec、退火56℃15sec、72℃延伸60sec/kb(35个循环);72℃延伸5min(1个循环)。overlap pcr扩增条件同pcr,按照等摩尔比添加不同的模板。
[0086]
2、培养基组成
[0087]
sc-ura缺陷液体培养基:20g/l葡萄糖,2g/l氨基酸混合物(不含尿嘧啶),6.7g/l不含氨基酸的酵母氮源;
[0088]
sc-ura缺陷固体培养基:20g/l葡萄糖,2g/l氨基酸混合物(不含尿嘧啶),6.7g/l不含氨基酸的酵母氮源,20g/l琼脂;
[0089]
ypd种子培养基:20g/l葡萄糖、20g/l蛋白胨、10g/l酵母浸粉,余量为水;
[0090]
ypd固体培养基:20g/l葡萄糖,20g/l蛋白胨,10g/l酵母提取物,20g/l琼脂;
[0091]
ypd发酵培养基:50g/l葡萄糖、20g/l蛋白胨、10g/l酵母浸粉,余量为水。
[0092]
3、发酵条件
[0093]
对照组:将重组菌株nhej-ly00和hr-ly00接入5ml种子培养基28℃,250转/分,培养24h,再将这5ml培养基转接到50ml新鲜的发酵培养基中,调整菌密度,使最终菌密度在od600nm下为0.2,28℃,250转/分,培养72h,取培养液用于产物分析。
[0094]
实验组:将经过三轮转化的高产番茄红素重组菌株接入5ml种子培养基28℃,250转/分,培养24h,再将这5ml培养基转接到50ml新鲜的发酵培养基中,调整菌密度,使最终菌密度在od600nm下为0.2,28℃,250转/分,培养72h,取培养液用于产物分析。
[0095]
4、荧光强度测量和番茄红素产物分析
[0096]
gfp荧光强度测量:用gfp报告基因转化的菌株在3ml sc-ura液体培养基中于28℃生长24h。以4500rpm/min离心2min后,转移中度培养液悬浮液放入含有200ul新鲜sc-ura液体培养基的96孔聚苯乙烯板(黑色板,透明底部)(corning incorporated 3603,美国)中,
最终od600nm为0.2。然后将细胞在28℃孵育48小时。取合适体积的培养液稀释至细胞od600nm在0.4至0.8之间。使用多模式酶标仪(spectramax m2,分子设备公司,美国)分析gfp荧光(激发:488nm,发射:530nm),并使用紫外分光光度计(tu-1810)测量细胞密度(od600)。通过荧光显微镜(olympus cx41,东京,日本)观察gfp荧光的图像。
[0097]
番茄红素的提取和分析:发酵72h后,分别取对照组和实验组500μl发酵液于2ml离心管中,12000rpm/min下离心5分钟,弃上清液。然后吸取1ml超纯水清洗细胞,吹吸均匀后,12000rpm/min下离心5分钟,重复操作2次。然后再加入1ml 3m hcl溶液,沸水浴3分钟,冰浴2min,重复3次后,12000rpm/min下离心5分钟,弃去上清液。然后吸取1ml超纯水清洗细胞,吹吸均匀后,12000rpm/min下离心5分钟,重复操作2次,去上清。然后加入含有0.1%bht(w/v)的丙酮中,震荡15min后,12000rpm/min下离心5分钟。最后用一次性医用注射器吸取上清液,经0.22μm有机滤膜过滤,使用配备bds hypersil c18色谱柱(4.6
×
150mm,5μm)和uv/vis检测器(waters 2489)的hplc系统(waters e2695)用于分析生产的类胡萝卜素。其中,检测器water 2489uv/vis在471nm处检测到番茄红素的信号,同时配制不同浓度梯度的番茄红素标品得到标准曲线,并对实验菌株的番茄红素谱图进行峰面积积分,根据标准曲线与样品的峰面积确定出实验菌株的番茄红素产量;波长设置为470nm;流动相:乙腈:甲醇:二氯甲烷(9:40:1);柱温设定为22℃,流速为0.3ml/min。
[0098]
5、结果
[0099]
在这项研究中,我们通过对解脂耶氏酵母非同源重组机制中的关键连接酶dl4及协同因子paxx和xrcc4的过表达,使得荧光强度提升了4.67倍;同时,截短ura3标签的启动子得到高压力筛选标签,荧光强度提升了22.74倍;采用优化后的多轮迭代转化流程,荧光强度再次提升了1.87倍。综合上述三方面的优化,在解脂耶氏酵母中成功建立了一种基于非同源末端连接机制的基因组整合方法。该方法实现了多个外源片段多拷贝随机地插入到基因组中。利用该方法能够将crte、crtb及crti三个外源片段同时整合到解脂耶氏酵母基因组中,得到一系列高产番茄红素产量的菌株。与依赖同源重组机制整合到rdna位点的菌株相比,利用基于非同源末端连接机制的基因组整合方法得到的菌株,番茄红素产量提升了23.88倍。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips