
 商标分类
商标分类  商标转让
商标转让 
一种邻位芳基取代的叔膦化合物的合成方法与流程
 2021-02-02 12:02:37|
2021-02-02 12:02:37| 297|
297| 起点商标网
起点商标网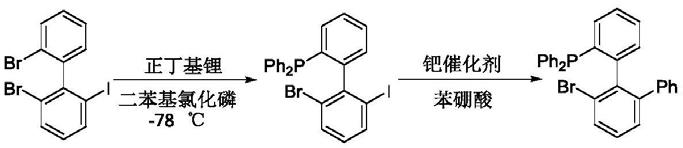
[0001]
本发明属于有机化学技术领域,涉及一种叔膦化合物及其合成方法,更具体地说,本发明涉及一种邻位芳基取代的叔膦化合物及其绿色经济的简单合成方法。
背景技术:
[0002]
叔膦化合物广泛应用于过渡金属催化的偶联反应当中,其对于提高催化剂活性、反应产率、区域选择性和对映选择性有着极为关键的促进作用。随着过渡金属催化偶联反应的快速发展,该类化合物的研究受到越来越多人关注,因而叔膦化合物的修饰也被广泛报道,但邻位芳基取代的叔膦化合物合成仍然存在很大挑战。
[0003]
已有报道的合成邻位芳基化叔膦化合物的文献中,主要包括高活性的金属锂试剂以及铑、钯、钌等金属催化,从而引入最关键的芳基取代基,该方法具有较大的局限性,存在反应试剂昂贵、底物范围窄、催化成本高、反应需要低/高温环境、反应条件缺乏绿色经济性、不能够大量制备等弊端。
[0004]
文献(eur.j.org.chem 2015,29,6515)中通过强碱正丁基锂与原料先发生一步锂卤交换,然后经过亲核取代以及钯催化suzuki偶联的方法多步合成了系列苯基修饰的单膦配体(如下反应式所示)。该合成路线中使用的正丁基锂危险性高且需-78℃超低温能耗高,不适合工业生产。
[0005][0006]
文献(chem.commun.2014,50,2193)以二价钯为催化剂,通过叔膦氧化物和三氟甲烷磺酸碘苯的交叉偶联再经还原后得到系列芳基修饰的叔膦化合物(如下反应式所示)。该路线通过c-h活化的方法实现了叔膦化合物的修饰,但反应底物需要前期氧化和后期还原,反应步骤繁琐,钯催化剂价格高昂,且底物范围有限。
[0007][0008]
文献(angew.chem.int.ed.2017,56,7233)以一价铑为催化剂,通过叔膦化合物和溴代芳烃的交叉偶联直接得到邻位芳基修饰的叔膦化合物,该路线虽然实现了叔膦的直接修饰(如下反应式所示),该方法使用的铑催化剂价格高昂,并且在体系中需要加入有机溶剂,缺乏绿色经济性。
[0009][0010]
文献(org.lett.2019,21,2885)以二价钌为催化剂,通过叔膦化合物和碘代芳烃的交叉偶联直接得到邻位芳基修饰的叔膦化合物,此合成路线实现了廉价钌催化的叔膦化合物的合成(如下反应式所示),但该方法使用较昂贵的高活性碘苯作为芳基化试剂,并且反应中仍需要加入有机溶剂,不符合绿色经济的要求。
[0011][0012]
综上所述,现有技术中邻位芳基修饰叔膦化合物的制备方法具有对反应条件要求高、安全风险大、操作复杂、底物范围有限、步骤繁琐、成本高、三废多、不适合工业化生产等缺点。
技术实现要素:
[0013]
有鉴于此,本申请公开了一种邻位芳基修饰叔膦化合物的绿色经济且简单制备方法,解决了现有技术中邻位芳基取代叔膦化合物的制备过程繁琐、生产成本高、操作工艺复杂、收率低、缺乏绿色经济性等问题。
[0014]
本发明提供了一种邻位芳基修饰叔膦化合物的绿色经济且简单的制备方法,其以联苯-2-二苯基膦1和氯代物2为原料,在钌催化剂、配体和无机碱存在下,无溶剂体系中加热反应得到芳基修饰叔膦化合物3,所述方法的合成路线如下式所示:
[0015][0016]
其中:r
1
选自氢、c1-c6烷基、c1-c4烷氧基、三氟甲基、羟基、酯基、萘基、氰基或卤素;r
2
选自氢、烷基、烷氧基、烷巯基、酯基、羟基、萘基、氰基或卤素;ar选自苯基、c1-c6烷基取代苯基、c1-c4烷氧取代苯基、三氟甲基苯基、卤素取代苯基、羰基取代苯基、醛基取代苯基、萘基、噻吩或吡啶。
[0017]
进一步地,在上述技术方案中,所述联苯-2-二苯基膦1与氯代物2摩尔比为1:1-20。
[0018]
进一步地,在上述技术方案中,所述配体选自丙氨酸、甘氨酸、苯丙氨酸、缬氨酸、亮氨酸、异亮氨酸、叔亮氨酸、n-boc-l-亮氨酸、n-boc-l-异亮氨酸、n-boc-l-叔亮氨酸、n-ac-异亮氨酸、boc-1-氨基环丙基甲酸、(2s,3s)-2-(boc-氨基)-3-甲基戊酸、n-boc-1-氨基
环丁烷羧酸、n-ac-d-缬氨酸、2-氨基异丁酸等氨基酸以及异戊酸、2-甲基丁酸、三甲基乙酸、三苯基乙酸、环己基乙酸、1-金刚烷乙酸、1-金刚烷甲酸、2,2-二苯基丙酸、2,4,6-三甲基苯甲酸、1,1-环丁烷二羧酸、2-金刚烷酮-5-甲酸或草酸。
[0019]
进一步地,在上述技术方案中,所述钌催化剂选自带有4-甲基异丙基苯基、三苯基膦、二甲基亚砜、乙腈、环辛二烯、五甲基环戊二烯、十二羰基或乙基环戊二烯配体配位的钌金属盐中的任意一种或者多种混合。
[0020]
进一步地,在上述技术方案中,所述无机碱选自醋酸钠、醋酸钾、醋酸铯、三氟乙酸锂、三氟乙酸钠、三氟乙酸钾、碳酸锂、碳酸钠、碳酸钾、碳酸铯、磷酸钾、磷酸二氢钾、磷酸氢二钾、氟化钾、碳酸氢钾、硫酸氢钾或叔丁醇钾。
[0021]
进一步地,在上述技术方案中,所述钌催化剂、配体、无机碱与联苯-2-二苯基膦1摩尔比为0.01-0.1:0.05-1:1-5:1。
[0022]
进一步地,在上述技术方案中,所述反应在惰性氛围中(例如氩气、氮气等)进行。
[0023]
进一步地,在上述技术方案中,所述加热反应温度为100-180℃。
[0024]
与现有技术相比,本发明涉及一种邻位芳基取代的叔膦化合物的绿色经济且简单的制备方法,其具有如下有益效果:
[0025]
1)本发明反应过程中,不需要加入有机溶剂,降低了反应成本,绿色经济;2)本发明合成步骤简单,只需一步完成,合成过程中不需要引入其他官能团或复杂的反应流程来构建c-c键,是一种简单的邻位芳基化叔膦化合物的修饰方法,且本发明合成工艺绿色经济、操作过程简单;3)本发明反应过程中所需要的原材料容易获得,产物收率良好(高达82%),且合成过程中对设备的要求低,具有一定的放大应用前景。
具体实施方式
[0026]
下面结合具体实施案例对本发明作进一步详细说明。本实施案例在本发明技术方案的前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施案例。
[0027]
实施例1
[0028]
本实施例的邻位芳基化叔膦化合物iii-1(4
″-
甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦,合成路线如下:
[0029][0030]
在反应釜中加入67.4mg(0.2mmol)2-二苯基膦-联苯(化合物1a)、500.0μl对氯苯甲醚(化合物2a,4.0mmol)、3.7mg对伞花烃二氯化钌二聚体、3.9mg叔亮氨酸和29.4mg醋酸钾,搅拌混合均匀后向反应体系中通氩气,控制反应在氩气氛围中、温度在140℃条件下反应24h后冷却至室温,硅藻土抽滤、减压蒸馏后柱层色谱分离得72.8mg产物3a,收率为82%。
1
h nmr(400mhz,cdcl
3
)δ7.40-7.35(m,2h),7.26-7.23(m,4h),7.19-7.14(m,8h),7.08(d,j=
8.8hz,2h),7.01(dd,j=7.6hz,3.2hz,1h),6.97(d,j=7.2hz,1h),6.84(t,j=7.2hz,2h),6.65(d j=8.4hz,2h),3.73(s,3h).化学位移δ7.40-7.35,多重峰,归属为苯环上的两个氢;化学位移δ7.26-7.23,多重峰,归属为苯环上的四个氢;化学位移δ7.19-7.14,多重峰,归属为苯环上的八个氢;化学位移δ7.08,两重峰,归属为苯环上的两个氢;化学位移δ7.01,四重峰,归属为苯环上的一个氢;化学位移δ6.97,二重峰,归属为苯环上的一个氢;化学位移δ6.84,三重峰,归属为苯环上的两个氢;化学位移δ6.65,二重峰,归属为苯环上的两个氢;化学位移δ3.73,单峰,归属为与苯基相连甲氧基的三个氢;
13
c nmr(100mhz,cdcl
3
)δ158.3,148.4(d,j=32.4hz),140.7(d,j=1.9hz),140.3(d,j=6.9hz),138.3(d,j=13.3hz),137.9(d,j=13.0hz),136.4(d,j=12.1hz),134.7(d,j=2.0hz),134.1(d,j=1.0hz),133.8(d,j=20.0hz),133.2(d,j=18.7hz),131.4(d,j=3.2hz),131.3(d,j=1.0hz),131.2(d,j=6.0hz),129.9,128.6,128.5,128.4,128.3,128.2(d,j=5.9hz),127.9,127.3,126.2,113.2,55.2.
31
p nmr(162mhz,cdcl
3
)δ-14.4.hrms(esi+)exact mass calculated for[m+h]
+
(c
31
h
25
op):445.1716,found:445.1705.
[0031]
综合上述核磁、质谱测试结果,可以确定,本实施例制得的目标化合物为(4
″-
甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦。
[0032]
实施例2
[0033]
本实施例的邻位芳基化叔膦化合物3b(4
″
,5-二甲氧基[1,1':2',1
″
三联苯基]-2-基)二苯基膦,合成路线如下:
[0034][0035]
上述所述邻位芳基化叔膦化合物3b采用如下方法制得,包括如下步骤:
[0036]
在反应釜中加入73.6mg(0.2mmol)(5-甲氧基-[1,1'-联苯]-2-基)二苯基膦(化合物1b)、500.0μl对氯苯甲醚、3.7mg对伞花烃二氯化钌二聚体、3.9mg叔亮氨酸和29.4mg醋酸钾,搅拌混合均匀后向反应体系中通氩气,控制反应在氩气氛围中、温度在140℃条件下反应24h后冷却至室温,硅藻土抽滤、减压蒸馏后柱层色谱分离得75.8mg产物3b,收率为80%。
1
h nmr(400mhz,cdcl
3
)δ7.40(d,j=4.0hz,2h),7.26-7.24(m,4h),7.20-7.14(m,5h),7.10(d,j=8.8hz,2h),7.05(d,j=7.2hz,1h),6.94(dd,j=8.8hz,j=8.0hz,1h),6.81(t,j=6.8hz,2h),6.76-6.73(m,2h),6.66(d,j=8.4hz,2h),3.75(s,3h),3.70(s,3h).
13
c nmr(100mhz,cdcl
3
)δ159.9,158.4,150.1(d,j=34.7hz),140.7(d,j=1.9hz),140.4(d,j=6.8hz),138.8(d,j=18.1hz),138.7(d,j=18.7hz),136.4(d,j=2.2hz),134.0(d,j=1.1hz),133.6,133.3(d,j=35.5hz),132.9,131.2,131.2,130.0,128.4,128.3,128.3,128.1(d,j=5.8hz),128.0,127.7,127.3(d,j=9.3hz),126.2,116.2(d,j=6.6hz),113.9,113.2,55.3,55.3.
31
p nmr(162mhz,cdcl
3
)δ-16.5.hrms(esi+)exact mass calculated for[m+h]
+
(c
32
h
27
o
2
p):475.1821,found:475.1810.
[0037]
实施例3
[0038]
本实施例的邻位芳基化叔膦化合物3c(3
″-
氯-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦,合成路线如下:
[0039][0040]
上述所述邻位芳基化叔膦化合物3c采用如下方法制得,包括如下步骤:
[0041]
在反应釜中加入67.4mg(0.2mmol)2-二苯基膦-联苯、520.0μl间二氯苯(化合物2c,4.0mmol)、3.7mg对伞花烃二氯化钌二聚体、3.9mg叔亮氨酸和29.4mg醋酸钾,搅拌混合均匀后向反应体系中通氩气,控制反应在氩气氛围中、温度在140℃条件下反应24h后冷却至室温,硅藻土抽滤、减压蒸馏后柱层色谱分离得60.0mg产物3c,收率为67%。
1
h nmr(400mhz,cdcl
3
)δ7.40-7.38(m,2h),7.28-7.21(m,9h),7.20-7.09(m,6h),7.04(d,j=4.8hz,2h),7.02-7.00(m,1h),6.94(d,j=7.6hz,1h),6.91(dd,j=7.2hz,1.6hz,1h).
13
c nmr(100mhz,cdcl
3
)δ147.5(d,j=32.1hz),143.5,140.3(d,j=6.7hz),140.0(d,j=1.2hz),137.9(d,j=12.9hz),137.4(d,j=12.3hz),136.5(d,j=12.2hz),134.5(d,j=1.9hz),134.0,133.8,133.5,133.2(d,j=18.8hz),131.5(d,j=3.4hz),131.1(d,j=5.8hz),130.2(d,j=2.2hz),129.8,128.8,128.6(d,j=3.3hz),128.4(d,j=4.3hz),128.4(d,j=3.1hz),128.3(d,j=1.1hz),128.2,128.0,127.6,127.0,126.6.
31
p nmr(162mhz,cdcl
3
)δ-14.2.hrms(esi
+
)exact mass calculated for[m+h]
+
(c
30
h
22
clp):449.1226,found:449.1211.
[0042]
实施例4
[0043]
采用2a对甲氧基氯苯为原料,通过改变反应原料1,采用实施例1类似的制备方法,制得了一系列邻位芳基修饰的叔膦衍生物;具体结果如下:
[0044]
邻位芳基化叔膦化合物3d(5-氯-4
″-
甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以5-氯-2-二苯基膦-联苯为原料,参照实施例1的方法,得到产物3d,收率为65%。
[0045][0046]
邻位芳基化叔膦化合物3e(4
″-
甲氧基-5-甲基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以5-甲基-2-二苯基膦-联苯为原料,参照实施例1的方法,得到产物3e,收率为78%。
[0047]
[0048]
邻位芳基化叔膦化合物3f(5-氟-4
″-
甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以5-氟-2-二苯基膦-联苯为原料,参照实施例1的方法,得到产物3f,收率为55%。
[0049][0050]
邻位芳基化叔膦化合物3g(4
″-
甲氧基-4-甲基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以4-甲基-2-二苯基膦-联苯为原料,参照实施例1的方法,得到产物3g,收率为78%。
[0051][0052]
邻位芳基化叔膦化合物3h(4
″-
甲氧基-4-三氟甲基-[1,1':2',1
″-
三联苯基])-2-基)二苯基膦(如下结构式所示)的制备方法,以4-三氟甲基-2-二苯基膦-联苯为原料,参照实施例1的方法,得到产物3h,收率为43%。
[0053][0054]
邻位芳基化叔膦化合物3i(4-氯-4
″-
甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以4-氯-2-二苯基膦-联苯为原料,参照实施例1的方法,得到产物3i,收率为67%。
[0055][0056]
邻位芳基化叔膦化合物3j(4
″-
甲氧基-4'-甲硫基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以(4'-甲硫基-[1,1'-联苯]-2-基)二苯基膦为原料,参照实施例1的方法,得到产物3j,收率为72%。
[0057][0058]
邻位芳基化叔膦化合物3k(4',4
″-
二甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯
基膦(如下结构式所示)的制备方法,以(4'-甲氧基-[1,1'-联苯]-2-基)二苯基膦为原料,参照实施例1的方法,得到产物3k,收率为67%。
[0059][0060]
邻位芳基化叔膦化合物3l(4'-氯-4
″-
甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以(4'-氯-[1,1'-联苯]-2-基)二苯基膦为原料,参照实施例1的方法,得到产物3l,收率为58%。
[0061][0062]
邻位芳基化叔膦化合物3m(4
″-
甲氧基-5'-甲基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以(3'-甲基-[1,1'-联苯]-2-基)二苯基膦为原料,参照实施例1的方法,得到产物3m,收率为79%。
[0063][0064]
实施例5
[0065]
采用1a 2-二苯基膦-联苯为原料,通过改变反应原料2,采用实施例1类似的制备方法,制得了一系列邻位芳基修饰的叔膦衍生物;具体结果如下:
[0066]
邻位芳基化叔膦化合物3n([1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以氯苯为原料,参照实施例1的方法,得到产物3n,收率为66%。
[0067][0068]
邻位芳基化叔膦化合物3o(2
″-
氟-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以邻氟氯苯为原料,参照实施例1的方法,得到产物3o,收率为53%。
[0069][0070]
邻位芳基化叔膦化合物3p 2
″-
(二苯基膦基)-[1,1':2',1
″-
三联苯基]-2-甲醛
(如下结构式所示)的制备方法,以邻氯苯甲醛为原料,参照实施例1的方法,得到产物3p,收率为46%。
[0071][0072]
邻位芳基化叔膦化合物3q(3
″-
氟-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以间氟氯苯为原料,参照实施例1的方法,得到产物3q,收率为49%。
[0073][0074]
邻位芳基化叔膦化合物3r(3
″-
甲基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以间氯甲苯为原料,参照实施例1的方法,得到产物3r,收率为71%。
[0075][0076]
邻位芳基化叔膦化合物3s(3
″-
甲氧基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以间氯苯甲醚为原料,参照实施例1的方法,得到产物3s,收率为80%。
[0077][0078]
邻位芳基化叔膦化合物3t(4
″-
氟-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以对氟氯苯为原料,参照实施例1的方法,得到产物3t,收率为64%。
[0079][0080]
邻位芳基化叔膦化合物3u(4
″-
甲基-[1,1':2',1
″-
三联苯基]-2-基)二苯基膦(如下结构式所示)的制备方法,以对氯甲苯为原料,参照实施例1的方法,得到产物3u,收率为71%。
[0081][0082]
邻位芳基化叔膦化合物3u 1-(2
″-
(二苯基膦基)-[1,1':2',1
″-
三联苯基]-4-基)乙-1-酮(如下结构式所示)的制备方法,以对氯苯乙酮为原料,参照实施例1的方法,得到产物3u,收率为78%。
[0083][0084]
邻位芳基化叔膦化合物3w 2-(2'-(二苯基膦基)-[1,1'-联苯]-2-基)吡啶(如下结构式所示)的制备方法,以2-氯吡啶为原料,参照实施例1的方法,得到产物3w,收率为38%。
[0085][0086]
本实施例邻位芳基化叔膦化合物3aa二苯基(2'-(噻吩-2-基)-[1,1'-联苯]-2-基)膦(如下结构式所示)的制备方法,以2-氯噻吩为原料,参照实施例1的方法,得到产物3aa,收率为55%。
[0087][0088]
从以上实施例以及测定的数据可以看出,采用本申请的方式制备邻位芳基化叔膦化合物的方法操作方便简单,所需要的原材料容易获得,反应无需加入额外有机溶剂,生产成本低,且在合成过程中对设备的要求低,适合工业化生产。
[0089]
以上仅是本发明的优选实施方式,应当指出的是,上述优选实施方式不应视为对本发明的限制,本发明的保护范围应当以权利要求所限定的范围为准。对于本技术领域的普通技术人员来说,在不脱离本发明的精神和范围内,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips