
 商标分类
商标分类  商标转让
商标转让 
基于全基因组选择烟气有害成分释放量预测方法及应用与流程
 2021-01-07 14:01:36|
2021-01-07 14:01:36| 406|
406| 起点商标网
起点商标网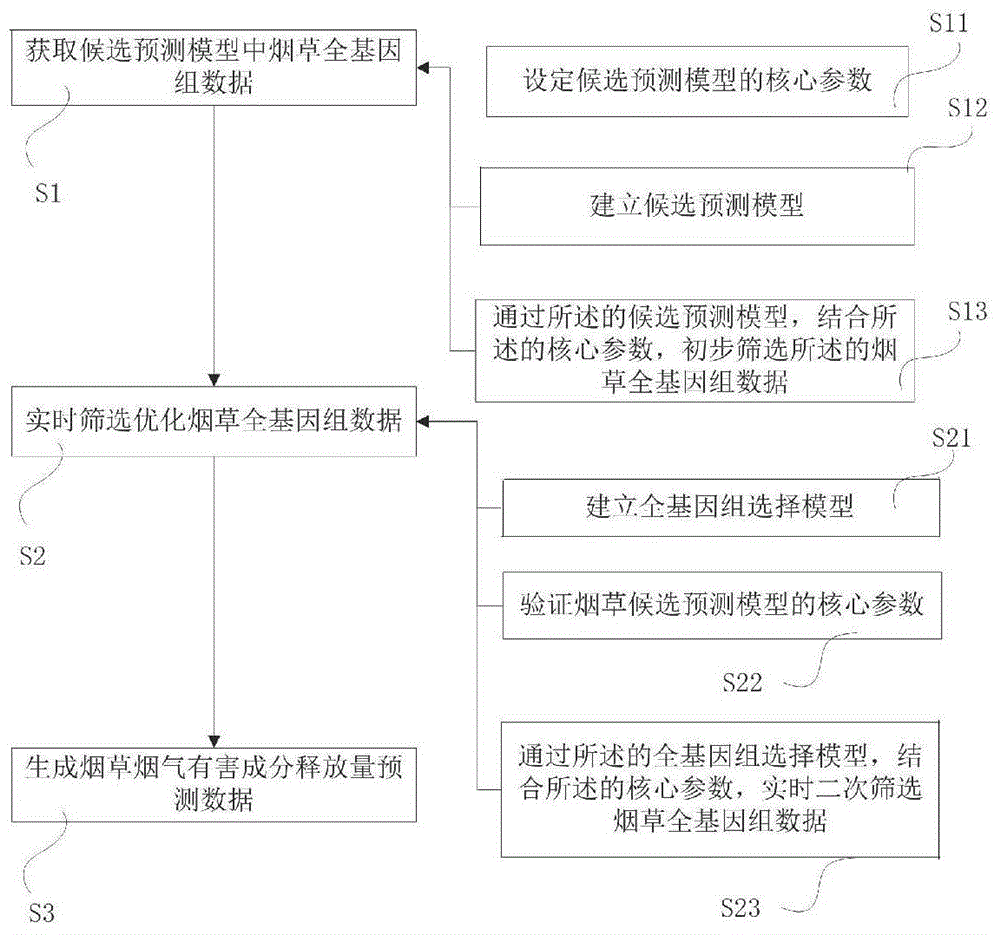
本发明属于生物技术领域,具体涉及一种基于全基因组选择烟气有害成分释放量预测方法及应用。
背景技术:
烟草(nicotianatabacuml.)是茄科烟草属一年生经济作物,因其能产生一种特有的植物碱-烟碱,且该物质能提供烟民吸食时获得极大的满足感和愉悦感而被广泛种植。但是,烟草又是一种备受争议的经济作物,因吸烟对人体健康无益,故此,培育低危害且优质的烟草品种就成为烟草新品种选育的一个重要方向。
卷烟主流烟气中有害成分性状是影响低危害烟草品种选育成败的核心因素。而卷烟烟气是一种极其复杂的混合物,它是在卷烟抽吸过程中由烟草燃烧、裂解和蒸馏而产生的。目前,已检出的烟气中化合物约7000种,有1000多种与烟叶的成分相同,表明烟叶中挥发性较高的香味物质和有害物质可以经过蒸馏直接影响烟气的质量,其余大部分是由热解和热解合成中产生的。
近年来,针对卷烟吸食(燃烧)时产生的主流烟气中有害成分释放量的测定十分繁杂、耗时耗工、效率低下、结果准确度低且不稳定。
传统模式,倘若获得相对准确可靠的卷烟主流烟气中有害成分释放量值,需经过一个完整而漫长的烟草田间生育期,成熟后需分期分批采摘叶片、烘烤、分级、叶片处理、切丝、前处理、烟支卷制后,进一步采用操作复杂、通量小的昂贵仪器设备检测卷烟主流烟气中的有害成分释放量,过程繁琐、效率低下且耗时较长。
技术实现要素:
针对以上目前对卷烟吸食(燃烧)时产生的主流烟气中有害成分释放量的测定十分繁杂、耗时耗工、效率低下、结果准确度低且不稳定的技术问题及缺陷,本发明提供一种基于全基因组选择烟气有害成分释放量预测方法及应用,实现基于烟草苗期(早期)的基因型数据,获得成熟烟草卷烟制品主流烟气有害成分释放量值数据。
本发明具体通过以下技术方案实现:一种基于全基因组选择烟气有害成分释放量预测方法,所述的方法具体包括如下步骤:获取候选预测模型中烟草全基因组数据;实时筛选优化烟草全基因组数据;生成烟草烟气有害成分释放量预测数据。
进一步地,于步骤获取候选预测模型中烟草全基因组数据之中,还包括如下步骤:设定候选预测模型的核心参数;建立候选预测模型;通过所述的候选预测模型,结合所述的核心参数,初步筛选所述的烟草全基因组数据。
进一步地,于步骤实时筛选优化烟草全基因组数据之中,还包括如下步骤:建立全基因组选择模型;验证烟草候选预测模型的核心参数;通过所述的全基因组选择模型,结合所述的核心参数,实时二次筛选烟草全基因组数据。
进一步地,所述的核心参数包括:分子标记数量、训练群体规模、训练群体与测试群体比例、模型预测精度值。
进一步地,所述的全基因组选择模型包括:苯并芘全基因组选择模型、4-甲基亚硝胺基-1-3-吡啶基-1-丁酮全基因组选择模型、氢氰酸全基因组选择模型、氨全基因组选择模型、巴豆醛全基因组选择模型、苯酚全基因组选择模型和一氧化碳全基因组选择模型。
进一步地,所述的烟草烟气具体为烟草卷烟制品的主流烟气;所述的烟草烟气有害成分释放量预测数据包括:苯并芘释放量数据、4-甲基亚硝胺基-1-3-吡啶基-1-丁酮释放量数据、氢氰酸释放量数据、氨释放量数据、巴豆醛释放量数据、苯酚释放量数据和一氧化碳释放量数据。
进一步地,所述的苯并芘释放量数据的计算公式为:
所述的4-甲基亚硝胺基-1-3-吡啶基-1-丁酮释放量数据的计算公式为:
所述的氢氰酸释放量数据的计算公式为:
所述的氨释放量数据的计算公式为:
所述的巴豆醛释放量数据的计算公式为:
所述的苯酚释放量数据的计算公式为:
所述的一氧化碳释放量数据的计算公式为:
其中,bayesab[a]p为苯并芘释放量的全基因组选择模型,b[a]p为苯并芘;bayesannk为4-甲基亚硝胺基-1-3-吡啶基-1-丁酮释放量的全基因组选择模型;nnk为4-甲基亚硝胺基-1-3-吡啶基-1-丁酮;bayesbhcn为氢氰酸释放量的全基因组选择模型;hcn为氢氰酸;bayescnh3为氨释放量的全基因组选择模型;nh3为氨;bayesbcro为巴豆醛释放量的全基因组选择模型;cro为巴豆醛;bayesbphe为苯酚释放量的全基因组选择模型;phe为苯酚;bayescco为一氧化碳释放量的全基因组选择模型;co为一氧化碳;bayesb为候选预测模型,bayesa为候选预测模型,bayesc为候选预测模型,n1为分子标记数量,n2为训练群体规模,n3为训练群体与测试群体比例,n4为模型预测精度值。
为实现上述目的,本发明还提供一种基于全基因组选择烟气有害成分释放量预测方法的应用,所述的基于全基因组选择烟气有害成分释放量预测方法应用于分析烟草群体苗期的基因型数据,并于全基因组范围内,预测出所述烟草群体中,各烟草植株成熟后制成卷烟制品中主流烟气有害成分释放量的表型值数据;
应用于分析烟草群体或烟草品种的基因型数据,通过在全基因组范围内,预测烟草成熟后制成卷烟制品中主流烟气有害成分释放量值数据,于烟草苗期时,获取所述烟草成熟后卷烟制品主流烟气中,有害成分释放量的表型值数据。
为实现上述目的,本发明还提供一种基于全基因组选择烟气有害成分释放量预测系统,所述的系统具体包括:获取单元,用于获取候选预测模型中烟草全基因组数据;筛选单元,用于实时筛选优化烟草全基因组数据;生成单元,用于生成烟草烟气有害成分释放量预测数据;
所述的获取单元,还包括:设定模块,用于设定候选预测模型的核心参数;第一建模模块,用于建立候选预测模型;第一筛选模块,用于通过所述的候选预测模型,结合所述的核心参数,初步筛选所述的烟草全基因组数据;
所述的筛选单元,还包括:第二建模模块,用于建立全基因组选择模型;验证模块,用于验证烟草候选预测模型的核心参数;第二筛选模块,通过所述的全基因组选择模型,结合所述的核心参数,实时二次筛选烟草全基因组数据。
为实现上述目的,本发明还提供一种基于全基因组选择烟气有害成分释放量预测平台,包括:处理器、存储器以及基于全基因组选择烟气有害成分释放量预测平台控制程序;其中,在所述的处理器执行所述的基于全基因组选择烟气有害成分释放量预测平台控制程序,所述的基于全基因组选择烟气有害成分释放量预测平台控制程序被存储在所述存储器中,所述的基于全基因组选择烟气有害成分释放量预测平台控制程序,实现所述的基于全基因组选择烟气有害成分释放量预测方法步骤。
为实现上述目的,本发明还提供一种计算机可读取存储介质,所述计算机可读取存储介质存储有基于全基因组选择烟气有害成分释放量预测平台控制程序,所述的基于全基因组选择烟气有害成分释放量预测平台控制程序,实现所述的基于全基因组选择烟气有害成分释放量预测方法步骤。
为实现上述目的,本发明还提供一种芯片系统,所述芯片系统包括至少一个处理器,当程序指令在所述至少一个处理器中执行时,使得所述芯片系统执行所述的基于全基因组选择烟气有害成分释放量预测方法步骤。
与现有技术相比,本发明具有以下有益效果:
本发明通过一种基于全基因组选择烟气有害成分释放量预测方法及应用、系统、平台、存储介质,可以实现通过烟草苗期(早期)的基因型数据,获得成熟烟草卷烟制品主流烟气有害成分释放量值数据,并且具备操作方便、快捷、高效、科学且结果精准可靠的特性。
也就是说,通过本发明可以实现利用烟草苗期(早期)的基因型数据来计算或模拟出2年后经过繁杂处理检测而获得的卷烟主流烟气中有害成分释放量值数据,通过本发明的模型或方法具有方便、快捷、高效、科学且结果精准可靠的特性。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明一种基于全基因组选择烟气有害成分释放量预测方法流程架构示意图;
图2为本发明之针对卷烟主流烟气中的有害成分苯并芘(b[a]p)释放量建立的全基因组选择模型bayesab[a]p示意图;
其中,图2中图(a)为分子标记数量(n1)对bayesab[a]p模型的b[a]p释放量预测精度影响示意图;图2中图(b)为训练群体规模(n2)对bayesab[a]p模型的b[a]p释放量预测精度影响示意图;图2中图(c)为训练群体与测试群体比例(n3)对bayesab[a]p模型的b[a]p释放量预测精度影响示意图;图2中图(d)为于bayesab[a]p模型中,不同候选预测模型对预测精度值(n4)的影响示意图;
图3为本发明之针对卷烟主流烟气中的4-甲基亚硝胺基-1-3-吡啶基-1-丁酮(nnk)释放量的全基因组选择模型bayesannk示意图;
其中,图3中图(a)为分子标记数量(n1)对bayesannk模型的nnk释放量预测精度影响示意图;图3中图(b)为训练群体规模(n2)对bayesannk模型的nnk释放量预测精度影响示意图;图3中图(c)为训练群体与测试群体比例(n3)对bayesannk模型的nnk释放量预测精度影响示意图;图3中图(d)为bayesannk模型中,不同候选预测模型对预测精度值(n4)的影响示意图;
图4为本发明之针对卷烟主流烟气中的氢氰酸(hcn)释放量的全基因组选择模型bayesbhcn示意图;
其中,图4中图(a)为分子标记数量(n1)对bayesbhcn模型的hcn释放量预测精度影响示意图;图4中图(b)为训练群体规模(n2)对bayesbhcn模型的hcn释放量预测精度影响示意图;图4中图(c)为训练群体与测试群体比例(n3)对bayesbhcn模型的hcn释放量预测精度影响示意图;图4中图(d)为bayesbhcn模型中,不同候选预测模型对预测精度值(n4)的影响示意图;
图5为本发明之针对卷烟主流烟气中的氨(nh3)释放量的全基因组选择模型bayescnh3示意图;
其中,图5中图(a)为分子标记数量(n1)对bayescnh3模型的nh3释放量预测精度影响示意图;图5中图(b)为训练群体规模(n2)对bayescnh3模型的nh3释放量预测精度影响示意图;图5中图(c)为训练群体与测试群体比例(n3)对bayescnh3模型的nh3释放量预测精度影响示意图;图5中图(d)为bayescnh3模型中,不同候选预测模型对预测精度值(n4)的影响示意图;
图6为本发明之针对卷烟主流烟气中的巴豆醛(cro)释放量的全基因组选择模型bayesbcro示意图;
其中,图6中图(a)为分子标记数量(n1)对bayesbcro模型的cro释放量预测精度影响示意图;图6中图(b)为训练群体规模(n2)对bayesbcro模型的cro释放量预测精度影响示意图;图6中图(c)为训练群体与测试群体比例(n3)对bayesbcro模型的cro释放量预测精度影响示意图;图6中图(d)为bayesbcro模型中,不同候选预测模型对预测精度值(n4)的影响示意图;
图7为本发明之针对卷烟主流烟气中的苯酚(phe)释放量的全基因组选择模型bayesbphe示意图;
其中,图7中图(a)为分子标记数量(n1)对bayesbphe模型的phe释放量预测精度影响示意图;图7中图(b)为训练群体规模(n2)对bayesbphe模型的phe释放量预测精度影响示意图;图7中图(c)为训练群体与测试群体比例(n3)对bayesbphe模型的phe释放量预测精度影响示意图;图7中图(d)为bayesbphe模型中,不同候选预测模型对预测精度值(n4)的影响示意图;
图8为本发明之针对卷烟主流烟气中的一氧化碳(co)释放量的全基因组选择模型bayescco示意图;
其中,图8中图(a)为分子标记数量(n1)对bayescco模型的co释放量预测精度影响示意图;图8中图(b)为训练群体规模(n2)对bayescco模型的co释放量预测精度影响示意图;图8中图(c)为训练群体与测试群体比例(n3)对bayescco模型的co释放量预测精度影响示意图;图8中图(d)为bayescco模型中,不同候选预测模型对预测精度值(n4)的影响示意图;
图9为本发明一种基于全基因组选择烟气有害成分释放量预测系统架构示意图;
图10为本发明一种基于全基因组选择烟气有害成分释放量预测平台架构示意图;
图11为本发明一种实施例中计算机可读取存储介质架构示意图;
本发明目的实现、功能特点及优点将结合实施例,参照附图做进一步说明。
具体实施方式
为便于更好的理解本发明的目的、技术方案和优点更加清楚,下面结合附图和具体的实施方式对本发明作进一步说明,本领域技术人员可由本说明书所揭示的内容轻易地了解本发明的其它优点与功效。
本发明亦可通过其它不同的具体实例加以施行或应用,本说明书中的各项细节亦可基于不同观点与应用,在不背离本发明的精神下进行各种修饰与变更。
需要说明,若本发明实施例中有涉及方向性指示(诸如上、下、左、右、前、后……),则该方向性指示仅用于解释在某一特定姿态(如附图所示)下各部件之间的相对位置关系、运动情况等,如果该特定姿态发生改变时,则该方向性指示也相应地随之改变。
另外,若本发明实施例中有涉及“第一”、“第二”等的描述,则该“第一”、“第二”等的描述仅用于描述目的,而不能理解为指示或暗示其相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。其次,各个实施例之间的技术方案可以相互结合,但是必须是以本领域普通技术人员能够实现为基础,当技术方案的结合出现相互矛盾或无法实现时,应当认为这种技术方案的结合不存在,也不在本发明要求的保护范围之内。
优选地,本发明一种基于全基因组选择烟气有害成分释放量预测方法应用在一个或者多个终端或者服务器中。所述终端是一种能够按照事先设定或存储的指令,自动进行数值计算和/或信息处理的设备,其硬件包括但不限于微处理器、专用集成电路(applicationspecificintegratedcircuit,asic)、可编程门阵列(field-programmablegatearray,fpga)、数字处理器(digitalsignalprocessor,dsp)、嵌入式设备等。
所述终端可以是桌上型计算机、笔记本、掌上电脑及云端服务器等计算设备。所述终端可以与客户通过键盘、鼠标、遥控器、触摸板或声控设备等方式进行人机交互。
本发明为实现一种基于全基因组选择烟气有害成分释放量预测方法、系统、平台及存储介质。如图1所示,是本发明实施例提供的基于全基因组选择烟气有害成分释放量预测方法的流程图。
在本实施例中,所述基于全基因组选择烟气有害成分释放量预测方法,可以应用于具备显示功能的终端或者固定终端中,所述终端并不限定于个人电脑、智能手机、平板电脑、安装有摄像头的台式机或一体机等。
所述基于全基因组选择烟气有害成分释放量预测方法也可以应用于由终端和通过网络与所述终端进行连接的服务器所构成的硬件环境中。网络包括但不限于:广域网、城域网或局域网。本发明实施例的基于全基因组选择烟气有害成分释放量预测方法可以由服务器来执行,也可以由终端来执行,还可以是由服务器和终端共同执行。
例如,对于需要进行基于全基因组选择烟气有害成分释放量预测终端,可以直接在终端上集成本发明的方法所提供的基于全基因组选择烟气有害成分释放量预测功能,或者安装用于实现本发明的方法的客户端。再如,本发明所提供的方法还可以软件开发工具包(softwaredevelopmentkit,sdk)的形式运行在服务器等设备上,以sdk的形式提供基于全基因组选择烟气有害成分释放量预测功能的接口,终端或其他设备通过所提供的接口即可实现基于全基因组选择烟气有害成分释放量预测功能。
以下结合附图对本发明作进一步阐述。
如图1所示,本发明提供了一种基于全基因组选择烟气有害成分释放量预测方法,所述的方法具体包括如下步骤:s1、获取候选预测模型中烟草全基因组数据;s2、实时筛选优化烟草全基因组数据;s3、生成烟草烟气有害成分释放量预测数据。
在本发明方案中,通过获取候选预测模型中烟草全基因组数据,并实时筛选优化烟草全基因组数据,最终生成烟草烟气有害成分释放量预测数据,所述方法可以实现通过烟草苗期(早期)的基因型数据,获得成熟烟草卷烟制品主流烟气有害成分释放量值数据,并且具备操作方便、快捷、高效、科学且结果精准可靠的特性。
也就是说,生成烟草烟气有害成分释放量预测数据,是经初步筛选获得的候选预测模型bayesa、bayesb和bayesc基础上,通过实验对卷烟制品主流烟气代表性有害成分(苯并芘(b[a]p)、nnk、氢氰酸(hcn)、氨(nh3)、巴豆醛(cro)、苯酚(phe)和一氧化碳(co))释放量表型值的预测结果进行不断筛选、优化、验证而获得,通过所述方法也分别获得了分子标记数量(n1)、训练群体规模(n2)、训练群体与测试群体比例(n3)及模型预测精度值(n4)等核心参数值。
具体地,于步骤获取候选预测模型中烟草全基因组数据之中,还包括如下步骤:s11、设定候选预测模型的核心参数;s12、建立候选预测模型;s13、通过所述的候选预测模型,结合所述的核心参数,初步筛选所述的烟草全基因组数据。
在本发明实施例中,通过对候选预测模型的核心参数进行明确设定,并建立候选预测模型,通过所述的候选预测模型,结合所述的核心参数,初步筛选所述的烟草全基因组数据。
也就是说,为使上述模型对卷烟制品主流烟气代表性有害成分释放量表型值的预测精度达到最优,对两类候选预测模型bayes(包含a、b和c三种)和rrblup的分子标记数量(n1)、训练群体规模(n2)、训练群体与测试群体比例(n3)及模型预测精度值(n4)等核心参数值进行了明确规定。
具体地,于步骤实时筛选优化烟草全基因组数据之中,还包括如下步骤:s21、建立全基因组选择模型;s22、验证烟草候选预测模型的核心参数;s23、通过所述的全基因组选择模型,结合所述的核心参数,实时二次筛选烟草全基因组数据。
在本发明实施例中,通过建立全基因组选择模型,即分别针对苯并芘(b[a]p)、nnk、氢氰酸(hcn)、氨(nh3)、巴豆醛(cro)、苯酚(phe)和一氧化碳(co)共7种,分别建立相对应的全基因组选择模型:bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco,再通过验证烟草候选预测模型的核心参数,通过所述的全基因组选择模型,结合所述的核心参数,实时二次筛选烟草全基因组数据,并对生成烟草烟气有害成分释放量预测数据进行不断筛选、优化、验证,而最终生成烟草烟气有害成分释放量预测数据,所述的烟草烟气具体为烟草卷烟制品的主流烟气。
具体地,所述的核心参数包括:分子标记数量、训练群体规模、训练群体与测试群体比例、模型预测精度值。
也就是说,在对烟草烟气有害成分释放量预测数据进行不断筛选、优化、验证时,通过本发明所述方法获得有分子标记数量(n1)、训练群体规模(n2)、训练群体与测试群体比例(n2)及模型预测精度值(n4)等核心参数值。
具体地,所述的全基因组选择模型包括:苯并芘全基因组选择模型、4-甲基亚硝胺基-1-3-吡啶基-1-丁酮全基因组选择模型、氢氰酸全基因组选择模型、氨全基因组选择模型、巴豆醛全基因组选择模型、苯酚全基因组选择模型和一氧化碳全基因组选择模型。
也就是说,在本发明方案中,分别针对苯并芘(b[a]p)、nnk、氢氰酸(hcn)、氨(nh3)、巴豆醛(cro)、苯酚(phe)和一氧化碳(co)共7种,分别建立相对应的全基因组选择模型:即分别为:苯并芘全基因组选择模型bayesab[a]p、4-甲基亚硝胺基-1-3-吡啶基-1-丁酮全基因组选择模型bayesannk、氢氰酸全基因组选择模型bayesbhcn、氨全基因组选择模型bayescnh3、巴豆醛全基因组选择模型bayesbcro、苯酚全基因组选择模型bayesbphe和一氧化碳全基因组选择模型bayescco。
具体地,所述的烟草烟气具体为烟草卷烟制品的主流烟气;所述的烟草烟气有害成分释放量预测数据包括:苯并芘释放量数据、4-甲基亚硝胺基-1-3-吡啶基-1-丁酮释放量数据、氢氰酸释放量数据、氨释放量数据、巴豆醛释放量数据、苯酚释放量数据和一氧化碳释放量数据。
较佳地,所述的苯并芘释放量数据的计算公式为:
所述的4-甲基亚硝胺基-1-3-吡啶基-1-丁酮释放量数据的计算公式为:
所述的氢氰酸释放量数据的计算公式为:
所述的氨释放量数据的计算公式为:
所述的巴豆醛释放量数据的计算公式为:
所述的苯酚释放量数据的计算公式为:
所述的一氧化碳释放量数据的计算公式为:
其中,bayesab[a]p为苯并芘释放量的全基因组选择模型,b[a]p为苯并芘;bayesannk为4-甲基亚硝胺基-1-3-吡啶基-1-丁酮释放量的全基因组选择模型;nnk为4-甲基亚硝胺基-1-3-吡啶基-1-丁酮;bayesbhcn为氢氰酸释放量的全基因组选择模型;hcn为氢氰酸;bayescnh3为氨释放量的全基因组选择模型;nh3为氨;bayesbcro为巴豆醛释放量的全基因组选择模型;cro为巴豆醛;bayesbphe为苯酚释放量的全基因组选择模型;phe为苯酚;bayescco为一氧化碳释放量的全基因组选择模型;co为一氧化碳;bayesb为候选预测模型,bayesa为候选预测模型,,bayesc为候选预测模型,n1为分子标记数量,n2为训练群体规模,n3为训练群体与测试群体比例,n4为模型预测精度值。
也就是说,预测烟草卷烟制品主流烟气中7种代表性有害成分释放量的全基因组选择模型分别为bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco,,所述模型的核心参数值分别为:
a)、针对苯并芘(b[a]p)释放量的全基因组选择模型bayesab[a]p:分子标记数量(n1)为4000个标记,即n1=4000个标记;训练群体规模(n2)为250个单株,即n2=250个单株;训练群体与测试群体比例(n3)为10:1,即n3=10:1(训练群体中的单株数量:测试群体中的单株数量=10:1);模型预测精度值(n4)为0.24,即n4=0.24。
b)、针对4-甲基亚硝胺基-1-3-吡啶基-1-丁酮(nnk)释放量的全基因组选择模型bayesannk:分子标记数量(n1)为4000个标记,即n1=4000个标记;训练群体规模(n2)为250个单株以上(含250个单株),即n2≥250个单株;训练群体与测试群体比例(n3)为1:5,即n3=1:5;模型预测精度值(n4)为0.03,即n4=0.03。
c)、针对氢氰酸(hcn)释放量的全基因组选择模型bayesbhcn:分子标记数量(n1)为1000个标记,即n1=1000个标记;训练群体规模(n2)为250个单株,即n2=250个单株;训练群体与测试群体比例(n3)为2:1,即n3=2:1;模型预测精度值(n4)为0.22,即n4=0.22。
d)、针对氨(nh3)释放量的全基因组选择模型bayescnh3:分子标记数量(n1)为22000个标记,即n1=22000个标记;训练群体规模(n2)为250个单株以上(含250个单株),即n2≥250个单株;训练群体与测试群体比例(n3)为4:1,即n3=4:1;模型预测精度值(n4)为0.09,即n4=0.09。
e)、针对巴豆醛(cro)释放量的全基因组选择模型bayesbcro:分子标记数量(n1)为1000个标记,即n1=1000个标记;训练群体规模(n2)为250个单株,即n2=250个单株;训练群体与测试群体比例(n3)为10:1,即n3=10:1;模型预测精度值(n4)为0.22,即n4=0.22。
f)、针对苯酚(phe)释放量的全基因组选择模型bayesbphe:分子标记数量(n1)为16000,即n1=16000个标记;训练群体规模(n2)为250个单株以上(包含250个单株),即n2≥250个单株;训练群体与测试群体比例(n3)为3:1,即n3=3:1;模型预测精度值(n4)为0.22,即n4=0.22。
g)、针对一氧化碳(co)释放量的全基因组选择模型bayescco:分子标记数量(n1)为1000个标记,即n1=1000个标记;训练群体规模(n2)为250个单株,即n2=250个单株;训练群体与测试群体比例(n3)为10:1,即n3=10:1;模型预测精度值(n4)为0.30,即n4=0.30。
换言之,通过本发明所述的方法,所述的预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型分别为:bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco,上述模型是在初步筛选获得的3种bayes候选预测模型(即bayesa、bayesb和bayesc)基础上,通过实验筛选、优化并验证获得各自核心参数分子标记数量(n1)、训练群体规模(n2)、训练群体与测试群体比例(n3)及模型预测精度值(n4)的具体数值。
所述的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco对应的4个核心参数值分别为:
分子标记数量(n1):a)针对苯并芘(b[a]p),n1=4000个标记;b)针对4-甲基亚硝胺基-1-3-吡啶基-1-丁酮(nnk),n1=4000个标记;c)针对氢氰酸(hcn),n1=1000个标记;d)针对氨(nh3),n1=22000个标记;e)针对巴豆醛(cro),n1=1000个标记;f)针对苯酚(phe),n1=16000个标记;g)针对一氧化碳(co),n1=1000个标记。
训练群体规模(n2):a)针对苯并芘(b[a]p),n2=250个单株;b)针对4-甲基亚硝胺基-1-3-吡啶基-1-丁酮(nnk),n2≥250个单株;c)针对氢氰酸(hcn),n2=250个单株;d)针对氨(nh3),n2≥250个单株;e)针对巴豆醛(cro),n2=250个单株;f)针对苯酚(phe),n2≥250个单株;g)针对一氧化碳(co),n2=250个单株。
训练群体与测试群体比例(n3):a)针对苯并芘(b[a]p),n3=10:1;b)针对4-甲基亚硝胺基-1-3-吡啶基-1-丁酮(nnk),n3=1:5;c)针对氢氰酸(hcn),n3=2:1;d)针对氨(nh3),n3=4:1;e)针对巴豆醛(cro),n3=10:1;f)针对苯酚(phe),n3=3:1;g)针对一氧化碳(co),n3=10:1。
模型预测精度值(n4):a)针对苯并芘(b[a]p),n4=0.24;b)针对4-甲基亚硝胺基-1-3-吡啶基-1-丁酮(nnk),n4=0.03;c)针对氢氰酸(hcn),n4=0.22;d)针对氨(nh3),n4=0.09;e)针对巴豆醛(cro),n4=0.22;f)针对苯酚(phe),n4=0.22;g)针对一氧化碳(co),n4=0.30。
下面以具体实施案例对本发明做进一步说明:
实施例1
预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco的构建及应用
一、实验材料
烤烟品种y3(低危害,危害性系数h=5.35)和烤烟品种k326(中高危害性,h=8.60)构建重组自交系(rils,f7)群体,该rils群体包含300个株系;此外,为构建具有烟草通用性的预测模型,组建了一个区别于双亲衍生群体的烟草自然群体,该自然群体由347份不同烟草品种(系)构成。
二、两种不同类型烟草群体的卷烟主流烟气中7种代表性有害成分释放量数据获得
试验材料成苗后移栽至大田,待大田烟草植株成熟后开始烟草叶片采收、烘烤、分级、切丝、检测、卷制、抽吸并统计主流烟气中7种代表性有害成分释放量值,其具体过程如下:
2.1仪器:直线吸烟机(英国斯茹林,sm450),近红外光谱仪(美国热电公司,antarisii)
2.2实验步骤:
2.2.1在自动吸烟机上抽吸卷烟,同时用玻璃纤维滤片烟气捕集器收集总粒相物。随机从实验室样品的每个包装中抽取尽量相等数量的卷烟,剔除有明显缺陷的烟支,在温度为22度,湿度为60%的平衡箱中放置48h,用于抽吸实验。在卷烟嘴端标准烟蒂长度处,用细软头笔划线,精确至0.5mm,划线时要小心,以免损伤烟支。划破、刺破及有缺陷的烟支均应弃去,用试料中的备用烟支代替。
2.2.2将已经在测试大气中调节至少12h的滤片放人滤片夹持器中,滤片粗糙的一面应面向进入的烟气,合上滤片夹持器,检查确认装配妥当。若烟气捕集器的设计中包含有孔垫片(垫圈),则将其嵌人,盖上密封装置(端帽)。
2.2.3直线吸烟机开机预热约20min之后,预热结束后,检查每通道的抽吸持续时间和抽吸频率应符合标准条件的规定。将烟气捕集器装到吸烟机上,使用皂膜流量计校正抽吸容量至35ml处,精确至±0.2ml,最小刻度0.1ml。
2.2.4将调节好的抽吸卷烟插人卷烟夹持器,使烟蒂末端正好接触捕集器内的有孔垫片。插入时应避免漏气或使烟支变形。有明显缺陷或插人时损伤的烟支均应弃去,并由调节好的备用烟支替代。确保卷烟位置正确,以使所有卷烟位置与孔道位置相一致。调整卷烟使燃烧锥到达烟蒂标记处时,启动抽吸终止装置。避免人为移动烟灰,以免干扰卷烟燃烧,应让烟灰自然落到烟灰盘上。当一支烟抽吸结束之后,立即插入新的卷烟,重复抽吸过程,直至将预定数量的卷烟抽吸完毕。
2.2.5抽吸结束后,取下烟气捕集器时,建议面向卷烟的一面朝下,以免卷烟夹持器上的污染物掉落到滤片上。
2.2.6近红外光谱仪预热3h以上,准直仪器,选择工作流,扣除空白背景。将剑桥滤片用镊子小心地取出,随即放入近红外样品杯中,置于中心位置。点击开始,检测样品的近红外光谱。
2.2.7基于云南中烟技术中心前期建立的烟气7项有害成分预测模型,对近红外光谱数据进行处理,得到样品烟气相关数据。
三、snp标记分析(以snp标记为例)
烟草基因组dna提取:采用常规ctab法或植物组织dna提取试剂盒均可,方法可参考已有的文献或试剂盒中的说明书。但需要对提取的烟草dna进行去除rna、蛋白质及其他有机杂质的纯化处理,使其达到开展snp芯片的要求;如若对烟草样品进行基因组重测序挖掘snp标记,则相应的烟草dna质量需按照测序公司的要求进行处理。
四、全基因组选择模型bayesab[a]p的构建及应用(以苯并芘(b[a]p)为例)
4.1候选预测模型的初步筛选
分别利用烟草重组自交系(rils,300个株系)、烟草自然群体(347个不同烟草品种)及两者混合后组成的烟草综合群体(647个株系)的各烟草株系snp基因型数据和主流烟气中苯并芘(b[a]p)释放量值,对r语言包中提供的4种原始模型rrblup、bayesa、bayesb和bayesc的基础参数(函数)进行优化。最终获得各候选模型的基础参数(函数)。
a)针对rrblup原始模型的基础函数:1.mixed.solve:把标记效应建模为随机效应或把行数据的基因型(genotypic)值用于a.mat函数(计算加性关系矩阵,预测育种值);2.kinship.blup:在基因型值预测中包含上位效应;3.gwa:关联映射;
具体参数设置:基因型数据填补、处理:a.mat(additiverelationshipmatrix)
impute<-a.mat(markers,max.missing=0.5;impute.method="em";n.core=4;return.imputed=t)
markers:基因型数据;max.missing:填补前允许最大的miss率,如果大于0.5,就删除所有样品此位点的snp;impute.method:填补方法;n.core:线程数;return.imputed:如果选择t,就返回填补后的数据。
训练集和测试集(可以根据实际需求比例设置,一般是91或82或64比例);train=as.matrix(sample(1:271,217));test<-setdiff(1:271,train);
在没要求设置训练集和测试集比例的分析统一按照8:2的比例进行(默认值);模型训练:mixed.solve(mixed-modelsolver)
height_answer<-mixed.solve(y=height,z=m_train,k=null,se=false,method="reml",return.hinv=false)
y:训练集表型数据;z:训练集基因型数据;k:协方差矩阵;se:标准差;method:模型训练方法
b)针对bayes原始模型的基础函数:数据输入与rrblup原始模型相同。
r语言包:bglr(bayesiangeneralizedlinearregression);eta<-list(list(x=markers,model='bayesb',probin=0.05));eta:两级列表,用于指定回归函数(或线性预测器);x:基因型文件;model:模型选择
模型训练:
system.time(fit_bb<-bglr(y=height,eta=eta,niter=10000,burnin=5000,thin=5,saveat=”,df0=5,s0=null,weights=null,r2=0.5))
y:表型文件;eta:两级列表,用于指定回归函数(或线性预测器)
niter,burnin,thin:(integer)thenumberofiterations(迭代次数),burn-inandthinning.
saveat:保护程序;r2:模型训练前的预期先验方差比例
4.2针对主流烟气中的苯并芘(b[a]p)模型bayesab[a]p的构建及应用
利用已确定基础参数的原始预测模型(即,候选预测模型),分别对含有300个、347个单株(系)的重组自交系群体和自然群体及其混合后产生的含有647个单株(系)的烟草综合群体开展针对卷烟主流烟气中苯并芘(b[a]p)释放量值预测的全基因组选择模型筛选、优化和验证。具体的方法如下:
首先,利用均匀分布于烟草全基因组上的50000个高质量snp标记分析含有647个单株(系)的烟草综合群体并获得基因型数据;其次,检测并统计上述烟草综合群体在烘烤后各单株(系)加工成卷烟的主流烟气中苯并芘(b[a]p)释放量数据,获得b[a]p表型数据;第三,以确定基础参数的2种类型4个候选预测模型(rrblup、bayesa、bayesb和bayesc)对上述烟草综合群体内各单株(系)的基因型数据和b[a]p释放量表型数据进行模拟,筛选、优化和验证预测模型中4个核心参数分子标记数量(n1)、训练群体规模(n2)、训练群体与测试群体比例(n3)和模型预测精度值(n4)的具体数值,最终构建预测卷烟主流烟气中苯并芘(b[a]p)释放量的全基因组选择模型bayesab[a]p。即,将50000个snp标记分为10个梯度(1000、2000、4000、7000、11000、16000、22000、29000、37000和50000)用于确定bayesab[a]p模型中的分子标记数量(n1)参数,结果见图2中的图(a);
将综合群体按照50、100、150、200、250和300个单株(系)作为训练群体,同时也将重组自交系群体(300个株系)、自然群体(347个株系)和综合群体(647个株系)分别单独作为训练群体用于确定bayesab[a]p模型中的训练群体规模(n2),其结果详见图2中图(b);
将综合群体内的647个株系按照训练群体中烟草植株数量:测试群体中烟草植株数量分别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1共11个梯度来确定bayesab[a]p模型中的训练群体与测试群体比例(n3),其结果见图2中的图(c);
将已获得具体核心参数(n1、n2和n3)的bayesab[a]p模型进行b[a]p表型值预测计算,获得主流烟气中的b[a]p释放量预测值,再与其各自的b[a]p真实检测值进行比较,获得预测精度,并确定最高预测精度值(n4)。最后,经上述实验验证、优化及实际应用后,构建获得预测卷烟主流烟气中苯并芘(b[a]p)释放量的全基因组选择模型bayesab[a]p,其公式如下:
其中,
具体地,图2是利用烟草综合群体(重组自交系群体和自然群体的混合群体)的基因型数据和实测表型数据并结合生物信息学,针对卷烟主流烟气中的有害成分苯并芘(b[a]p)释放量建立的全基因组选择模型bayesab[a]p。
其中,图2中图(a)为分子标记数量(n1)对bayesab[a]p模型的b[a]p释放量预测精度影响:横坐标为分子标记数量;纵坐标为bayesab[a]p模型对b[a]p释放量的预测精度;图中横坐标显示的1k、2k、4k、7k、11k、16k、22k、29k、37k和all分别表示用于烟草综合群体进行基因型分析的snp标记数量为1000、2000、4000、7000、11000、16000、22000、29000、37000和50000个。
图2中图(b)为训练群体规模(n2)对bayesab[a]p模型的b[a]p释放量预测精度影响:横坐标为训练群体规模(训练群体包含的烟草植株数量);纵坐标为bayesab[a]p模型对b[a]p释放量的预测精度。
图2中图(c)为训练群体与测试群体比例(n3)对bayesab[a]p模型的b[a]p释放量预测精度影响:横坐标为训练群体与测试群体的比例(即,训练群体中的烟草植株数量:测试群体中的烟草植株数量),1/5、1/4、1/3、1/2、1、2、3、4、5、6和10分别表示训练群体:测试群体的比值为别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1;纵坐标为bayesab[a]p模型对b[a]p释放量的预测精度。
图2中图(d)为不同候选预测模型对预测精度值(n4)的影响:横坐标表示r语言包中提供的4种原始模型:bayesa、bayesb、bayesc和rrblup;纵坐标表示该原始模型对待测烟草群体的b[a]p释放量预测的精度。
同理,按照建立苯并芘(b[a]p)全基因组选择模型:
类似方法,建立卷烟主流烟气中其余6种代表性有害成分(nnk、hcn、nh3、cro、phe和co)的全基因组选择模型,具体模型分别如下:
针对nnk释放量,其全基因组选择模型为:
具体地,针对卷烟主流烟气中的4-甲基亚硝胺基-1-3-吡啶基-1-丁酮(nnk)释放量的全基因组选择模型bayesannk,如图3所示,相应的:
其中,图3中图(a)为分子标记数量(n1)对bayesannk模型的nnk释放量预测精度影响:横坐标为分子标记数量;纵坐标为bayesannk模型对nnk释放量的预测精度;图中横坐标显示的1k、2k、4k、7k、11k、16k、22k、29k、37k和all分别表示用于烟草综合群体进行基因型分析的snp标记数量为1000、2000、4000、7000、11000、16000、22000、29000、37000和50000个。
图3中图(b)为训练群体规模(n2)对bayesannk模型的nnk释放量预测精度影响:横坐标为训练群体规模(训练群体包含的烟草植株数量);纵坐标为bayesannk模型对nnk释放量的预测精度。
图3中图(c)为训练群体与测试群体比例(n3)对bayesannk模型的nnk释放量预测精度影响:横坐标为训练群体与测试群体的比例(即,训练群体中的烟草植株数量:测试群体中的烟草植株数量),1/5、1/4、1/3、1/2、1、2、3、4、5、6和10分别表示训练群体:测试群体的比值为别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1;纵坐标为bayesannk模型对nnk释放量的预测精度。
图3中图(d)为不同候选预测模型对预测精度值(n4)的影响:横坐标表示r语言包中提供的4种原始模型:bayesa、bayesb、bayesc和rrblup;纵坐标表示该原始模型对待测烟草群体的nnk释放量预测的精度。
针对hcn释放量,其全基因组选择模型为:
具体地,针对卷烟主流烟气中的氢氰酸(hcn)释放量的全基因组选择模型bayesbhcn,如图4所示,相应的:
其中,图4中图(a)为分子标记数量(n1)对bayesbhcn模型的hcn释放量预测精度影响:横坐标为分子标记数量;纵坐标为bayesbhcn模型对hcn释放量的预测精度;图中横坐标显示的1k、2k、4k、7k、11k、16k、22k、29k、37k和all分别表示用于烟草综合群体进行基因型分析的snp标记数量为1000、2000、4000、7000、11000、16000、22000、29000、37000和50000个。
图4中图(b)为训练群体规模(n2)对bayesbhcn模型的hcn释放量预测精度影响:横坐标为训练群体规模(训练群体包含的烟草植株数量);纵坐标为bayesbhcn模型对hcn释放量的预测精度。
图4中图(c)为训练群体与测试群体比例(n3)对bayesbhcn模型的hcn释放量预测精度影响:横坐标为训练群体与测试群体的比例(即,训练群体中的烟草植株数量:测试群体中的烟草植株数量),1/5、1/4、1/3、1/2、1、2、3、4、5、6和10分别表示训练群体:测试群体的比值为别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1;纵坐标为bayesbhcn模型对hcn释放量的预测精度。
图4中图(d)为不同候选预测模型对预测精度值(n4)的影响:横坐标表示r语言包中提供的4种原始模型:bayesa、bayesb、bayesc和rrblup;纵坐标表示该原始模型对待测烟草群体的hcn释放量预测的精度。
针对nh3释放量,其全基因组选择模型为:
具体地,针对卷烟主流烟气中的氨(nh3)释放量的全基因组选择模型bayescnh3,如图5所示,相应的:
其中,图5中图(a)为分子标记数量(n1)对bayescnh3模型的nh3释放量预测精度影响:横坐标为分子标记数量;纵坐标为bayescnh3模型对nh3释放量的预测精度;图中横坐标显示的1k、2k、4k、7k、11k、16k、22k、29k、37k和all分别表示用于烟草综合群体进行基因型分析的snp标记数量为1000、2000、4000、7000、11000、16000、22000、29000、37000和50000个。
图5中图(b)为训练群体规模(n2)对bayescnh3模型的nh3释放量预测精度影响:横坐标为训练群体规模(训练群体包含的烟草植株数量);纵坐标为bayescnh3模型对nh3释放量的预测精度。
图5中图(c)为训练群体与测试群体比例(n3)对bayescnh3模型的nh3释放量预测精度影响:横坐标为训练群体与测试群体的比例(即,训练群体中的烟草植株数量:测试群体中的烟草植株数量),1/5、1/4、1/3、1/2、1、2、3、4、5、6和10分别表示训练群体:测试群体的比值为别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1;纵坐标为bayescnh3模型对nh3释放量的预测精度。
图5中图(d)为不同候选预测模型对预测精度值(n4)的影响:横坐标表示r语言包中提供的4种原始模型:bayesa、bayesb、bayesc和rrblup;纵坐标表示该原始模型对待测烟草群体的nh3释放量预测的精度。
针对cro释放量,其全基因组选择模型为:
具体地,针对卷烟主流烟气中的巴豆醛(cro)释放量的全基因组选择模型bayesbcro,如图6所示,相应的:
其中,图6中图(a)为分子标记数量(n1)对bayesbcro模型的cro释放量预测精度影响:横坐标为分子标记数量;纵坐标为bayesbcro模型对cro释放量的预测精度;图中横坐标显示的1k、2k、4k、7k、11k、16k、22k、29k、37k和all分别表示用于烟草综合群体进行基因型分析的snp标记数量为1000、2000、4000、7000、11000、16000、22000、29000、37000和50000个。
图6中图(b)为训练群体规模(n2)对bayesbcro模型的cro释放量预测精度影响:横坐标为训练群体规模(训练群体包含的烟草植株数量);纵坐标为bayesbcro模型对cro释放量的预测精度。
图6中图(c)为训练群体与测试群体比例(n3)对bayesbcro模型的cro释放量预测精度影响:横坐标为训练群体与测试群体的比例(即,训练群体中的烟草植株数量:测试群体中的烟草植株数量),1/5、1/4、1/3、1/2、1、2、3、4、5、6和10分别表示训练群体:测试群体的比值为别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1;纵坐标为bayesbcro模型对cro释放量的预测精度。
图6中图(d)为不同候选预测模型对预测精度值(n4)的影响:横坐标表示r语言包中提供的4种原始模型:bayesa、bayesb、bayesc和rrblup;纵坐标表示该原始模型对待测烟草群体的cro释放量预测的精度。
针对phe释放量,其全基因组选择模型为:
具体地,针对卷烟主流烟气中的苯酚(phe)释放量的全基因组选择模型bayesbphe,如图7所示,相应的:
其中,图7中图(a)为分子标记数量(n1)对bayesbphe模型的phe释放量预测精度影响:横坐标为分子标记数量;纵坐标为bayesbphe模型对phe释放量的预测精度;图中横坐标显示的1k、2k、4k、7k、11k、16k、22k、29k、37k和all分别表示用于烟草综合群体进行基因型分析的snp标记数量为1000、2000、4000、7000、11000、16000、22000、29000、37000和50000个。
图7中图(b)为训练群体规模(n2)对bayesbphe模型的phe释放量预测精度影响:横坐标为训练群体规模(训练群体包含的烟草植株数量);纵坐标为bayesbphe模型对phe释放量的预测精度。
图7中图(c)为训练群体与测试群体比例(n3)对bayesbphe模型的phe释放量预测精度影响:横坐标为训练群体与测试群体的比例(即,训练群体中的烟草植株数量:测试群体中的烟草植株数量),1/5、1/4、1/3、1/2、1、2、3、4、5、6和10分别表示训练群体:测试群体的比值为别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1;纵坐标为bayesbphe模型对phe释放量的预测精度。
图7中图(d)为不同候选预测模型对预测精度值(n4)的影响:横坐标表示r语言包中提供的4种原始模型:bayesa、bayesb、bayesc和rrblup;纵坐标表示该原始模型对待测烟草群体的phe释放量预测的精度。
针对co释放量,其全基因组选择模型为:
具体地,针对卷烟主流烟气中的一氧化碳(co)释放量的全基因组选择模型bayescco,如图8所示,相应的:
其中,图8中图(a)为分子标记数量(n1)对bayescco模型的co释放量预测精度影响:横坐标为分子标记数量;纵坐标为bayescco模型对co释放量的预测精度;图中横坐标显示的1k、2k、4k、7k、11k、16k、22k、29k、37k和all分别表示用于烟草综合群体进行基因型分析的snp标记数量为1000、2000、4000、7000、11000、16000、22000、29000、37000和50000个。
图8中图(b)为训练群体规模(n2)对bayescco模型的co释放量预测精度影响:横坐标为训练群体规模(训练群体包含的烟草植株数量);纵坐标为bayescco模型对co释放量的预测精度。
图8中图(c)为训练群体与测试群体比例(n3)对bayescco模型的co释放量预测精度影响:横坐标为训练群体与测试群体的比例(即,训练群体中的烟草植株数量:测试群体中的烟草植株数量),1/5、1/4、1/3、1/2、1、2、3、4、5、6和10分别表示训练群体:测试群体的比值为别为1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、6:1和10:1;纵坐标为bayescco模型对co释放量的预测精度。
图8中图(d)为不同候选预测模型对预测精度值(n4)的影响:横坐标表示r语言包中提供的4种原始模型:bayesa、bayesb、bayesc和rrblup;纵坐标表示该原始模型对待测烟草群体的co释放量预测的精度。
较佳地,于公式(1)、(2)、(3)、(4)、(5)、(6)、(7)中,bayesab[a]p为苯并芘释放量的全基因组选择模型,b[a]p为苯并芘;bayesannk为4-甲基亚硝胺基-1-3-吡啶基-1-丁酮释放量的全基因组选择模型;nnk为4-甲基亚硝胺基-1-3-吡啶基-1-丁酮;bayesbhcn为氢氰酸释放量的全基因组选择模型;hcn为氢氰酸;bayescnh3为氨释放量的全基因组选择模型;nh3为氨;bayesbcro为巴豆醛释放量的全基因组选择模型;cro为巴豆醛;bayesbphe为苯酚释放量的全基因组选择模型;phe为苯酚;bayescco为一氧化碳释放量的全基因组选择模型;co为一氧化碳;bayesb为候选预测模型,bayesa为候选预测模型,,bayesc为候选预测模型,n1为分子标记数量,n2为训练群体规模,n3为训练群体与测试群体比例,n4为模型预测精度值。
为实现上述目的,本发明还提供一种基于全基因组选择烟气有害成分释放量预测方法的应用,所述的基于全基因组选择烟气有害成分释放量预测方法应用于分析烟草群体苗期的基因型数据,并于全基因组范围内,预测出所述烟草群体中,各烟草植株成熟后制成卷烟制品中主流烟气有害成分释放量的表型值数据;
应用于分析烟草群体或烟草品种的基因型数据,通过在全基因组范围内,预测烟草成熟后制成卷烟制品中主流烟气有害成分释放量值数据,于烟草苗期时,获取所述烟草成熟后卷烟制品主流烟气中,有害成分释放量的表型值数据。
也就是说,所述的预测卷烟主流烟气代表性有害成分释放量的全基因组选择模型的应用,是利用所述的7个全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco分析烟草群体苗期的基因型数据并在全基因组范围内精准预测出该群体中各烟草植株成熟后制成卷烟制品中7种主流烟气有害成分释放量的表型值。
较佳地,预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型的应用,分别利用全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco分析烟草群体或烟草品种(系)的基因型数据,在全基因组范围内精准预测烟草成熟后制成卷烟制品中7种主流烟气有害成分释放量值,从而实现在烟草苗期(早期)就能获得其成熟后卷烟制成品的主流烟气中7种代表性有害成分释放量的精准表型值。
即所述的7个全基因组选择模型,在苗期(早期)的烟草群体或品种(系)中分析各植株基因型数据从而预测获得卷烟主流烟气中7种代表性有害成分释放量的应用。
所述的预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型的应用是利用bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco模型分析苗期(早期)待检测的烟草群体或品种(系)基因型值预测获得待测烟草成熟后,经采摘、烘烤、分级、切丝、卷制等一系列繁杂耗时工序而成为卷烟制成品的主流烟气中的苯并芘(b[a]p)、nnk、氢氰酸(hcn)、氨(nh3)、巴豆醛(cro)、苯酚(phe)和一氧化碳(co)释放量值。
综上所述,本发明的第一目的在于提供一种准确预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco;第二目的在于利用所述的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco在烟草群体或品种(系)中分析其早期(苗期)基因型数据从而准确预测获得卷烟制成品的主流烟气中7种代表性有害成分释放量值的应用。
本发明的第一目的是这样实现的,所述的预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco,其各自核心参数值n1、n2、n3和n4有明确的界定。
本发明的第二目的是这样实现的,所述的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco在烟草群体或品种(系)中分析各植株早期(苗期)基因型数据从而准确预测获得卷烟制成品的主流烟气中7种代表性有害成分释放量的应用。
为了科学、高效、精准的选择主流烟气中具有不同有害成分释放量水平的烟草品种,有针对性和特异性的选择具有低危害的后代烟草材料,本发明提供一种预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco,利用上述模型分别对重组自交系群体(rils)、自然群体及两者混合后的综合群体进行早期基因型数据和烟草制成品(卷烟)主流烟气中7种代表性有害成分释放量值采集、分析,在初选获得的bayesa、bayesb和bayesc候选预测模型基础上,筛选、优化并验证各模型中的分子标记数量(n1)、训练群体规模(n2)、训练群体与测试群体比例(n3)及模型预测精度值(n4)等4个核心参数。上述模型的最终建立,可用于全基因组范围内对卷烟主流烟气中7种代表性有害成分性状基因/qtl位点的辅助选择,以提高分子标记辅助选择的效率及低危害烟草品种选育的效率。
本发明一方面利用烤烟品种y3和k326(构建重组自交系(rils,f7)群体,同时,也组建了一个含有347份不同烟草品种(系)的自然群体,利用上述两个群体来代表全部的烟草群体或品种(系);另一方面采用经初选获得的3种候选预测模型bayesa、bayesb和bayesc,进一步筛选、优化并构建预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco,加速全基因组范围内分子标记选择在低危害烟草品种中的选育工作。
本发明所述的预测卷烟主流烟气中7种代表性有害成分释放量的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco具有科学、高效、准确和低成本的特点,可在培育低危害优良烟草新品种(系)中应用。
为实现上述目的,本发明还提供一种基于全基因组选择烟气有害成分释放量预测系统,如图9所示,所述的系统具体包括:获取单元,用于获取候选预测模型中烟草全基因组数据;筛选单元,用于实时筛选优化烟草全基因组数据;生成单元,用于生成烟草烟气有害成分释放量预测数据;
所述的获取单元,还包括:设定模块,用于设定候选预测模型的核心参数;第一建模模块,用于建立候选预测模型;第一筛选模块,用于通过所述的候选预测模型,结合所述的核心参数,初步筛选所述的烟草全基因组数据;
所述的筛选单元,还包括:第二建模模块,用于建立全基因组选择模型;验证模块,用于验证烟草候选预测模型的核心参数;第二筛选模块,通过所述的全基因组选择模型,结合所述的核心参数,实时二次筛选烟草全基因组数据。
在本发明系统方案实施例中,所述的基于全基因组选择烟气有害成分释放量预测系统中涉及的预测卷烟主流烟气中代表性有害成分释放量的全基因组选择模型及其应用、方法相同,此处不再赘述。
为实现上述目的,本发明还提供一种基于全基因组选择烟气有害成分释放量预测平台,如图10所示,包括:处理器、存储器以及基于全基因组选择烟气有害成分释放量预测平台控制程序;其中,在所述的处理器执行所述的基于全基因组选择烟气有害成分释放量预测平台控制程序,所述的基于全基因组选择烟气有害成分释放量预测平台控制程序被存储在所述存储器中,所述的基于全基因组选择烟气有害成分释放量预测平台控制程序,实现所述的基于全基因组选择烟气有害成分释放量预测方法步骤,步骤具体细节已在上文阐述,此处不再赘述。
为实现上述目的,本发明还提供一种计算机可读取存储介质,如图11所示,所述计算机可读取存储介质存储有基于全基因组选择烟气有害成分释放量预测平台控制程序,所述的基于全基因组选择烟气有害成分释放量预测平台控制程序,实现所述的基于全基因组选择烟气有害成分释放量预测方法步骤,步骤具体细节已在上文阐述,此处不再赘述。
为实现上述目的,本发明还提供一种芯片系统,所述芯片系统包括至少一个处理器,当程序指令在所述至少一个处理器中执行时,使得所述芯片系统执行所述的基于全基因组选择烟气有害成分释放量预测方法步骤,步骤具体细节已在上文阐述,此处不再赘述。
本发明通过一种基于全基因组选择烟气有害成分释放量预测方法及应用、系统、平台、存储介质,可以实现通过烟草苗期(早期)的基因型数据,获得成熟烟草卷烟制品主流烟气有害成分释放量值数据,并且具备操作方便、快捷、高效、科学且结果精准可靠的特性。
也就是说,通过本发明可以实现利用烟草苗期(早期)的基因型数据来计算或模拟出2年后经过繁杂处理检测而获得的卷烟主流烟气中有害成分释放量值数据,通过本发明的模型或方法具有方便、快捷、高效、科学且结果精准可靠的特性。
换言之,本发明一方面利用烤烟品种y3和烤烟品种k326构建基于双亲衍生的重组自交系(rils,f7)群体,此外,也组建了一个含有347份不同烟草品种(系)的自然群体,两种不同类型群体来代表全部烟草群体或烟草品种(系);另一方面采用初步筛选获得的bayesa、bayesb和bayesc候选预测模型,结合上述烟草材料的实测表型值,进一步筛选、优化并构建预测卷烟主流烟气中苯并芘(b[a]p)、nnk、氢氰酸(hcn)、氨(nh3)、巴豆醛(cro)、苯酚(phe)和一氧化碳(co)共7种代表性有害成分释放量的全基因组选择模型bayesab[a]p、bayesannk、bayesbhcn、bayescnh3、bayesbcro、bayesbphe和bayescco,加速全基因组范围内分子标记选择在主流烟气中具有低危害成分释放量的烟草品种选育工作,从而实现科学、高效、精准、可靠的培育出具有低危害烟草优良品种。即本发明所述的卷烟主流烟气7种代表性有害成分释放量的全基因组选择模型可以依据烟草群体苗期的基因型数据来精确的预测出所述群体中各植株成熟后制成卷烟制品中7种主流烟气有害成分释放量值,从而实现低危害烟草良种(系)的培育。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
因此,本发明专利的保护范围应以所附权利要求为准。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



