
 商标分类
商标分类  商标转让
商标转让 
一种高效合成1,6-二烯-3-酮衍生物的方法与流程
 2021-02-02 12:02:44|
2021-02-02 12:02:44| 297|
297| 起点商标网
起点商标网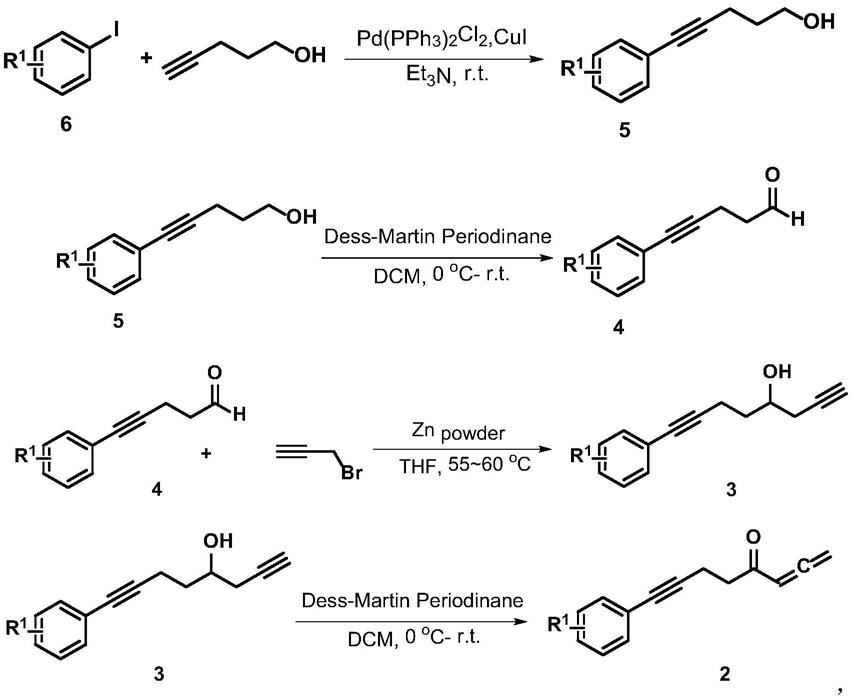
[0001]
本发明属于有机合成技术领域,具体涉及一种高效合成1,6-二烯-3-酮衍生物的方法。
背景技术:
[0002]
1,6-二烯-3-酮是一种重要的有机合成中间体,由于产品骨架的特殊性,不仅在医药方面表现出丰富的药理活性,而且所合成出的新材料性能突出,可替代多种进口材料,如感光材料、电荷输送材料、半导体粘合。另外,在微电子、有机发光半导体、光学设备等方面具有广泛的应用前景。
[0003]
目前已经有很多关于合成1,6-二烯-3-酮衍生物的文献报道,但现有合成方法都存在反应条件苛刻、操作复杂、污染环境等缺陷,要获得新的1,6-二烯-3-酮衍生物很困难。
技术实现要素:
[0004]
本发明的目的是提供一种高效合成1,6-二烯-3-酮衍生物的方法,操作简单,反应条件温和,原子利用率高。
[0005]
为实现上述目的,本发明提供一种高效合成1,6-二烯-3-酮衍生物的方法,在有机溶剂体系中,以式2化合物为原料,在75~85℃条件下搅拌回流反应,tlc跟踪反应至完全,反应液后处理得到式1化合物;
[0006]
其中式2化合物的结构式为式1化合物的结构式为其中,r
1
选自氢、c1-c5烷基、c1-c5烷氧基、酯基、卤素、噻吩基、吡啶基、三氟甲基、三甲基硅基中的一种。
[0007]
优选的,所述的r
1
选自氢(h)、甲基(me)、乙基(et)、正丙基(n-pr)、叔丁基(t-bu)、甲氧基(ome)、乙酯基(coome)、氟(f)、氯(cl)、溴(br)、噻吩基、吡啶基(py)、三氟甲基(cf
3
)、三甲基硅基(si(ch
3
)
3
)中的一种。
[0008]
优选的,所述有机溶剂选自甲苯、苯、二甲苯、均三甲苯、氯苯、二苯醚、乙腈、乙酸乙酯、甲醇、二氯甲烷、二氯乙烷、吡啶、1,4-二氧六环、四氢呋喃、n,n-二甲基甲酰胺、二甲亚砜中的一种或几种。
[0009]
进一步地,所述式2化合物通过以下步骤制备得到:
[0010]
1)在惰性气氛中,式6化合物与4-戊炔醇在双(三苯基膦)二氯化钯和碘化亚酮催化下,三乙胺为溶剂,室温反应,tlc跟踪反应至完全,乙酸乙酯萃取,干燥,抽滤,浓缩,柱层
析分离得到式5化合物;
[0011]
2)将式5化合物溶于二氯甲烷溶剂中,0℃缓慢加入戴斯马丁氧化剂固体颗粒,将温度升至室温继续反应2小时,抽滤除去固体,滤液用饱和碳酸氢钠水溶液淬灭,二氯甲烷萃取,干燥,抽滤,浓缩,柱层析分离得到式4化合物;
[0012]
3)将活化后的锌粉溶于四氢呋喃溶液中,搅拌状态下加入式4化合物,缓慢加入3-溴丙炔,55~60℃反应48~72小时,冷却至室温,加饱和氯化铵水溶液淬灭,乙酸乙酯萃取,干燥,抽滤,浓缩,柱层析分离得到式3化合物;
[0013]
4)将式3化合物溶于二氯甲烷溶剂中,0℃缓慢加入戴斯马丁氧化剂固体颗粒,将温度升至室温继续反应2小时,抽滤除去固体,滤液用饱和碳酸氢钠水溶液淬灭,二氯甲烷萃取,干燥,抽滤,浓缩,柱层析分离得到式2化合物;
[0014]
上述步骤1)至4)的反应路线及式2~式6化合物的结构式为:
[0015][0016][0017]
式中r
1
选自氢、c1-c5烷基、c1-c5烷氧基、酯基、卤素、噻吩基、吡啶基、三氟甲基、三甲基硅基中的一种。
[0018]
优选的,步骤1)中,所述式6化合物与4-戊炔醇的摩尔比为1.1:1。
[0019]
优选的,步骤2)中,所述式5化合物与戴斯马丁氧化剂的摩尔比为1:1.1。
[0020]
优选的,步骤3)中,所述式4化合物与3-溴丙炔的摩尔比为1:2。
[0021]
优选的,步骤4)中,所述式3化合物与戴斯马丁氧化剂的摩尔比为1:1.1。
[0022]
本发明通过串联反应首次设计合成了一系列底物联烯-7-炔-4-酮,该底物是一种反应位点多、官能团活性高的合成子,无需任何催化剂或氧化剂,仅在溶剂中便可顺利进行合成得到1,6-二烯-3-酮衍生物,该反应具有操作简单、反应条件温和、产物收率高、结构新
颖等优点。
具体实施方式
[0023]
下面结合具体实施例对本发明作进一步详细说明。
[0024]
下述实施例中,除非另有说明,所述的实验方法通常按照常规条件或制造厂商建议的条件实施;所述的原料、试剂均可通过市售购买的方式获得。
[0025]
实施例1:制备化合物1a
[0026]
双(三苯基膦)二氯化钯(0.35g,0.5mmol)和碘化亚酮(0.19g,1mmol)加入到500ml规格的史莱克瓶中,氩气氛围下依次加入溶剂三乙胺(200ml)、碘苯(11.22g,55mmol)和4-戊炔醇(4.2g,50mmol),室温反应12小时。乙酸乙酯萃取,无水硫酸镁干燥,抽滤,真空减压浓缩,洗脱剂为乙酸乙酯:石油醚=1:3,柱层析分离得到4-戊炔-1-醇(7.6g,95%)。
[0027]
4-戊炔-1-醇(7.6g,47.5mmol)溶于二氯甲烷(50ml)溶剂中,0℃缓慢加入戴斯马丁(22.2g,52.3mmol)氧化剂固体颗粒,将温度升至室温继续反应2小时。抽滤除去固体,滤液用饱和碳酸氢钠水溶液淬灭,二氯甲烷萃取,无水硫酸镁干燥,抽滤,真空减压浓缩,通过硅胶柱层析法,洗脱剂为乙酸乙酯:石油醚=1:30,得到苯基戊炔醛(7.1g,95%)。
[0028]
首先将活化后的锌粉(8.8g,134.7mmol)溶于四氢呋喃(150ml)溶液中,搅拌状态下加入苯基戊炔醛(7.1g,44.9mmol),缓慢加入3-溴丙炔(10.6g,89.8mmol),55~60℃反应48~72小时。冷却至室温,加饱和氯化铵水溶液淬灭,乙酸乙酯萃取,无水硫酸镁干燥,抽滤,真空减压浓缩,通过硅胶柱层析法,洗脱剂乙酸乙酯/石油醚先用1:30,后提高至1:10,得到1,7-二炔-4-醇(8.0g,90%)。
[0029]
将1,7-二炔-4-醇(8.0g,40.4mmol)溶于二氯甲烷(40ml)溶剂中,0℃缓慢加入戴斯马丁(18.8g,44.4mmol)氧化剂固体颗粒,将温度升至室温继续反应2小时。抽滤除去固体,滤液用饱和碳酸氢钠水溶液淬灭,二氯甲烷萃取,无水硫酸镁干燥,抽滤,真空减压浓缩,通过硅胶柱层析法,洗脱剂为乙酸乙酯:石油醚=1:30,得到联烯-7-炔-4-酮2a(6.8g,86%)。
[0030]
联烯-7-炔-4-酮2a(6.8g,34.7mmol)溶于甲苯(70ml)溶剂中,升温至80℃,反应48小时左右。真空减压浓缩,硅胶柱层析分离,洗脱剂为乙酸乙酯:石油醚=1:5,得到1,6-二烯-3-酮化合物1a(5.1g,75%)。
[0031]
本实施例中底物2a的结构表征数据如下:
1
h nmr(400mhz,cdcl
3
;δ,ppm):7.40-7.36(m,2h),7.28(s,3h),5.86-5.81(m,1h),5.29(d,j=7.2hz,2h),2.97-2.92(m,2h),2.73-2.67(m,2h).
13
c nmr(100mhz,cdcl
3
;δ,ppm):216.9,198.4,131.6,128.2,127.8,123.7,96.6,88.6,81.0,79.9,38.2,14.4.
[0032]
产物1a的结构表征数据如下:
1
h nmr(400mhz,dmso;δ,ppm):7.51-7.46(m,4h),7.44-7.40(m,1h),5.70(s,1h),3.47-3.44(m,2h),3.01-2.96(m,2h),2.51(d,j=2.8hz,2h).
13
c nmr(100mhz,dmso;δ,ppm):197.4,160.8,144.3,139.5,133.7,129.8,129.4,127.6,113.4,36.4,35.6,21.9.
[0033]
实施例1-14的反应合成路线如下所示:
[0034][0035][0036]
反应原料、反应条件及产率如表1所示:
[0037]
表1 1,6-二烯-3-酮类化合物的收率
[0038]
[0039]
[0040][0041]
*各步骤反应条件(包括投料比、催化剂、氧化剂、溶剂、温度和时间)不变,仅改变r
1
取代基。
[0042]
产物1b的结构表征数据如下:
1
h nmr(400mhz,dmso;δ,ppm):7.38(d,j=8.0hz,2h),7.29(d,j=8.0hz,2h),5.66(s,1h),3.42-3.39(m,2h),2.96-2.91(m,2h),2.51(d,j=7.2hz,2h),2.35(s,3h).
13
c nmr(100mhz,dmso;δ,ppm):197.3,161.0,144.4,139.8,138.4,131.0,130.0,127.6,112.9,36.4,35.5,21.8,21.6.
[0043]
产物1f的结构表征数据如下:
1
h nmr(400mhz,dmso;δ,ppm):7.44(d,j=8.8hz,2h),7.04(d,j=8.8hz,2h),5.62(s,1h),3.82(s,3h),3.40(d,j=2.0hz,2h),2.92(s,2h),2.50(d,j=7.2hz,2h).
13
c nmr(100mhz,dmso;δ,ppm):197.2,161.2,160.7,144.4,136.8,129.4,126.5,115.0,112.1,55.8,36.4,35.6,21.7.
[0044]
产物1i的结构表征数据如下:
1
h nmr(400mhz,dmso;δ,ppm):7.56-7.47(m,4h),5.71(s,1h),3.45(s,2h),3.00-2.93(m,2h),2.54(d,j=7.2hz,2h).
13
c nmr(100mhz,dmso;δ,ppm):197.3,160.4,142.9,140.2,134.2,132.5,129.5,129.2,113.8,36.3,35.6,21.9.
[0045]
产物1j的结构表征数据如下:
1
h nmr(400mhz,dmso;δ,ppm):7.67(d,j=8.4hz,2h),7.43(d,j=8.0hz,2h),5.72(s,1h),3.46-3.43(m,2h),2.99-2.93(m,2h),2.53(d,j=7.6hz,2h).
13
c nmr(100mhz,dmso;δ,ppm):197.3,160.4,143.0,140.4,132.8,132.4,129.4,123.0,113.8,36.3,35.6,21.9.
[0046]
本发明提供的产物结构中六元并四元环骨架可以开环,产物结构进一步衍生化,进而发挥药理活性,丰富药物分子库。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips