
 商标分类
商标分类  商标转让
商标转让 
一种3-溴甲基-4-溴苯乙酮的合成方法与流程
 2021-02-02 11:02:44|
2021-02-02 11:02:44| 483|
483| 起点商标网
起点商标网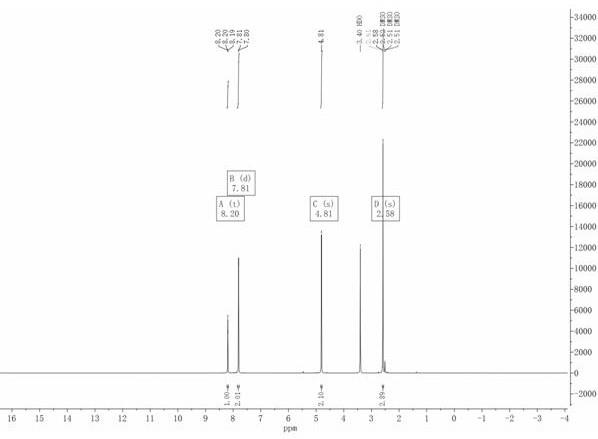
[0001]
本发明涉及有机合成技术领域,具体涉及一种3-溴甲基-4-溴苯乙酮的合成方法。
背景技术:
[0002]
3-溴甲基-4-溴苯乙酮,分子式c
9
h
8
br
2
o,相对分子质量291.97,外观为白色晶体,其结构式如下:丙型肝炎病毒(hcv)具有传染性强、传播途径复杂、流行面广、发病率较高等特点。hcv感染严重危害人类健康,目前,全球约有1.85亿人感染,感染率约3%,此外,每年新发hcv感染病例约有300-400万例左右。hcv是黄病毒科的一种正链rna病毒,因此,以ns5a为靶点的研究成为目前研究热点。维帕他韦(velpatasvir,cas:1377049-84-7)是吉利德公司开发的一种泛基因型ns5a抑制剂,对所有基因型hcv以及常见的ns5a突变和耐药性突变均有良好的体外抗病毒活性,市场前景广阔。3-溴甲基-4-溴苯乙酮是合成维帕他韦的中间体之一,开发出一条适合工业化生产的合成路线极具商业价值和社会意义。
[0003]
目前合成方法以下3类方法为主:1,专利文献wo2015191437a1报道路线如下,以2-溴-3-碘-苯甲醇为原料,先保护羟基,再上格氏反应上羰甲基,脱保护后取代即得目标化合物-;此路线四步,路线较长,原料不易得,成本较高。
[0004]
2,专利文献cn17629029a1公开方法为,以邻甲基苯胺为原料,经氨基乙酰化保护,傅克酰基化,脱氨基保护重氮化,最后溴代制得3-溴甲基4-溴苯乙酮,路线如下:此路线长,产率低,用溴化亚铜与重氮盐反应,工业化污染大,且不安全。
[0005]
3,专利文献cn17629029a1报道为,以3-甲基-4-溴苯乙酸为原料,先与韦伯胺缩合,在于甲基格氏试剂反应得到3-甲基-4-溴苯乙酮,最后溴代得到3-溴甲基4-溴苯乙酮;
此路线用韦伯胺和缩合试剂,价格较贵,且最后一步羰基α位和甲基均可溴代,且易多取代,选择性不好,收率低下,总成本高。
[0006]
综上所述,需求一种经济、安全的工业化工艺是很有必要的。
技术实现要素:
[0007]
本发明目的在于提供3-氯甲基-4-溴苯乙酮合成新方法,以解决上述问题。本发明以邻溴苯甲醛为原料经过酰化和卤化两步得到目标化合物-,选择性高、步骤少,收率高,且经济、安全,具体技术方案的步骤如下:本发明实施例提供了一种3-溴甲基-4-溴苯乙酮的合成方法,步骤如下:(1)化合物-的合成:邻溴苯甲醛在催化剂c1的作用下与乙酰胺发生酰化反应,得到化合物-。其中,反应温度为0-80℃,邻溴苯甲醛与催化剂c1的摩尔比为1:1.0-2.0,邻溴苯甲醛与乙酰胺的重量体积比为1:0.28-5.0g/ml,催化剂c1选自三溴氧磷、三氯氧磷、二溴亚砜、二氯亚砜和二硫酰氯等中的一种。
[0008]
(2)化合物-的合成:化合物-在催化剂c2的作用下与烷基卤硅烷试剂于溶剂s2中反应得到化合物-。其中,反应温度为0-110℃,化合物-、催化剂c2和烷基卤硅烷的摩尔比为1:0.5-2.0:1.0-5.0,催化剂c2为聚甲基氢硅氧烷或1,1,3,3-四甲基二硅氧烷等,x为cl、br或i中的一种。相应地,化合物-为以下结构中的一种:即化合物-为:3-氯甲基-4-溴苯乙酮(
ⅲ-
a)、3-溴甲基-4-溴苯乙酮(
ⅲ-
b)和3-碘甲基-4-溴苯乙酮(
ⅲ-
c)中的一种。
[0009]
其中,步骤(1)具体包括:在0-35℃条件下,将邻溴苯甲醛和乙酰胺溶解在溶剂s1中,在0-80℃条件下,滴加催化剂c1,滴加完成后,于0-80℃条件下保温反应,反应完全后加水(优选为冰水)中止反应,后处理得到化合物-。
[0010]
其中,在步骤(1)中,溶剂s1选自乙酰胺、二氯甲烷、1,2-二氯乙烷、石油醚、环己烷和乙腈等中的一种;优选为乙酰胺或二氯甲烷。
[0011]
优选地,在步骤(1)中,反应温度为20-25℃。
[0012]
其中,后处理过程包括:加入萃取剂进行萃取,分液后,有机相水洗,分液,有机相减压蒸去溶剂,加入乙酸乙酯:石油醚=1:5的溶剂重结晶,得到化合物-。
[0013]
其中,步骤(2)具体包括:在0-35℃条件下,将化合物-加入至溶剂s2中,在0-110℃条件下,依次滴加烷基卤硅烷试剂和催化剂c2,滴加完成后,于0-110℃条件下保温反应,反应完全后得到化合物-。
[0014]
其中,在步骤(2)中,溶剂s2选自二氯甲烷、1,2-二氯乙烷、氯苯、苯、甲苯、二甲苯、三氟乙酸和乙腈等中的一种;优选为二氯甲烷或乙腈中的一种。
[0015]
优选地,在步骤(2)中,催化剂c2为1,1,3,3-四甲基二硅氧烷。
[0016]
优选地,在步骤(2)中,反应温度为30-40℃。
[0017]
其中,在步骤(2)中,烷基卤硅烷试剂的结构为:;r
1
、r
2
、r
3
选自甲基、乙基、丙基和丁基等中的一种,x为cl、br或i中的一种。
[0018]
优选地,在步骤(2)中,烷基卤硅烷试剂为三甲氯硅烷、三甲基溴硅烷或三甲基碘硅烷。
[0019]
具体地,本发明提供的合成方法的步骤如下:(1)在0-35℃条件下,将邻溴苯甲醛和乙酰胺溶解在溶剂s1中,在0-80℃条件下,滴加催化剂c1,滴加完成后,于0-80℃条件下保温反应,反应完全后加水中止反应,后处理得到化合物-。
[0020]
其中,邻溴苯甲醛与催化剂c1的摩尔比为1:1.0-2.0,邻溴苯甲醛与乙酰胺的重量体积比为1:0.28-5.0g/ml,催化剂c1选自三溴氧磷、三氯氧磷、二溴亚砜、二氯亚砜和二硫酰氯等中的一种,溶剂s1选自乙酰胺、二氯甲烷、1,2-二氯乙烷、石油醚、环己烷和乙腈等中的一种。
[0021]
(2)在0-35℃条件下,将化合物-加入至溶剂s2中,在0-110℃条件下,依次滴加烷基卤硅烷试剂和催化剂c2,滴加完成后,于0-110℃条件下保温反应,反应完全后得到化合物-。
[0022]
其中,化合物-、催化剂c2和烷基卤硅烷的摩尔比为1:0.5-2.0:1.0-5.0,催化剂c2为聚甲基氢硅氧烷或1,1,3,3-四甲基二硅氧烷等;溶剂s2选自二氯甲烷、1,2-二氯乙烷、氯苯、苯、甲苯、二甲苯、三氟乙酸和乙腈等中的一种;烷基卤硅烷试剂的结构为:,r
1
、r
2
、r
3
为甲基、乙基、丙基或丁基等中的一种,x为cl、br或i中的一种。
[0023]
优选地,本发明提供的步骤如下:(1)在室温下,将邻溴苯甲醛和乙酰胺溶解在溶剂s1中,在15-25℃条件下,滴加二氯亚砜,滴加完成后,于20-25℃条件下保温反应,反应完全后加冰水中止反应,后处理得到化合物-。其中,邻溴苯甲醛与二氯亚砜的摩尔比为1:1.0-2.0,邻溴苯甲醛与乙酰胺的重量体积比为1:0.5-2.0g/ml,溶剂s1为乙酰胺或二氯甲烷。
[0024]
(2)在室温下,将化合物-加入至溶剂s2中,在20-40℃条件下,依次滴加烷基卤硅
烷试剂和1,1,3,3-四甲基二硅氧烷,滴加完成后,于30-40℃条件下保温反应,反应完全后得到化合物-。其中,化合物-、1,1,3,3-四甲基二硅氧烷和烷基卤硅烷的摩尔比为1:0.5-2.0:1.0-5.0,溶剂s2为二氯甲烷或乙腈;烷基卤硅烷试剂为三甲基氯硅烷、三甲基溴硅烷或三甲基碘硅烷。
[0025]
经检测,本发明仅需两步反应,反应条件温和,总收率可到60%,纯度可达99%以上。
附图说明
[0026]
图1是实施例4得到的3-溴甲基-4-溴苯乙酮的h-nmr图谱。
具体实施方式
[0027]
为使本发明的目的、技术方案和优点更加清楚,下面对本发明实施方式作进一步地详细描述。
[0028]
实施例1化合物-的合成室温(20-25℃)下,将185.02g邻溴苯甲醛溶解在185ml乙酰胺中,控温15-25℃下,滴加催化剂154.66g二氯亚砜,滴完,保温20-25℃反应,液相监控反应,约10-13小时反应完全,反应完毕后,控温0-30℃滴加冰水300g破坏,加入600ml二氯乙烷萃取,分液后,有机相200ml水洗1次,分液,有机相减压蒸去二氯甲烷,加入乙酸乙酯:石油醚=1:5的溶剂重结晶,得到产物190.5g浅黄色固体,含量99.62%,收率83.58%。
[0029]
实施例2化合物-的合成室温(20-25℃)下,将185.02g邻溴苯甲醛和70.89g乙酰胺溶解在555ml的二氯甲烷中,控温15-25℃下,滴加催化剂154.66g二氯亚砜,滴完,保温20-25℃反应,液相监控反应,约10-13小时反应完全,反应完毕后,控温0-30℃滴加冰水300g破坏,分液后,有机相200ml水洗1次,分液,有机相减压蒸去二氯甲烷,加入乙酸乙酯:石油醚=1:5的溶剂重结晶,得到产物178.87g浅黄色固体,含量99.47%,收率78.36%。
[0030]
实施例3化合物
ⅲ-
a的合成室温(20-25℃)下,将113.53g化合物-加入至568ml二氯甲烷中溶解,控温20-40℃下,依次滴加65.18g三甲基氯硅烷,67.16g1,1,3,3-四甲基二硅氧烷,加毕30-40℃反应,液相监控反应,至化合物-为0-3%,终止反应,降温,0-20℃加入300ml水搅拌30分钟,分液,有机相水洗两次,每次150ml,减压回收溶剂,浓干物加入乙酸乙酯:石油醚=1:2.5的溶剂重结晶,抽滤,烘干即得100.68g白色晶体化合物
ⅲ-
a,含量99.30%,收率80.78%,化合物
ⅲ-
a,1hnmr(400mhz,cdcl3)δ8.05 (s,1h),7.75 (d,j=8.4hz,1h), 4.73(s,2h),2.60(s,3h)。
[0031]
实施例4化合物
ⅲ-
b的合成室温(20-25℃)下,将113.53g化合物-加入至568ml二氯甲烷中溶解,控温20-40℃下,依次滴加91.85g三甲基溴硅烷,67.16g1,1,3,3-四甲基二硅氧烷,加毕30-40℃反应,液相监控反应,至化合物-为0-3%,终止反应,降温,0-20℃加入300ml水搅拌30分钟,分液,有机
相水洗两次,每次150ml,减压回收溶剂,浓干物加入乙酸乙酯:石油醚=1:2的溶剂重结晶,抽滤,烘干即得125.67g白色晶体化合物
ⅲ-
b,含量99.54%,收率85.69%,熔点:99-101℃。
1
h nmr(400mhz,dmso-d6)δ8.20 (t,j =1.3hz,1h), 7.81 (d,j = 1.3hz,2h), 4.81 (s,2h),2.58(s,3h)。
[0032]
实施例5化合物
ⅲ-
b的合成室温(20-25℃)下,将113.53g化合物-加入至300ml乙腈中溶解,控温20-40℃下,依次滴加91.85g三甲基溴硅烷,67.16g1,1,3,3-四甲基二硅氧烷,加毕30-40℃反应,液相监控反应,至化合物-为0-3%,终止反应,降温,0-20℃缓慢滴加60ml水搅拌30分钟,降温至-5~0℃结晶,搅拌2h,抽滤,烘干即得110.88g白色晶体化合物
ⅲ-
b,含量99.85%,收率75.84%,熔点:99-101℃。
[0033]
实施例6
ⅲ-
c的合成室温(20-25℃)下,将113.53g化合物-加入至568ml二氯甲烷中溶解,控温20-40℃下,依次滴加110.05g三甲基碘硅烷,67.16g1,1,3,3-四甲基二硅氧烷,加毕30-40℃反应,液相监控反应,至化合物-为0-3%,终止反应,降温,0-20℃加入300ml水搅拌30分钟,分液,有机相水洗两次,每次150ml,减压回收溶剂,浓干物加入乙酸乙酯:石油醚=1:1.5的溶剂重结晶,抽滤,烘干即得142.01g白色到浅黄色晶体化合物
ⅲ-
c,含量99.63%,收率83.48%。
[0034]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips