
 商标分类
商标分类  商标转让
商标转让 
VEGFR-2抑制剂的筛选方法、VEGFR-2抑制剂和抗肿瘤药物与流程
 2021-02-02 10:02:35|
2021-02-02 10:02:35| 313|
313| 起点商标网
起点商标网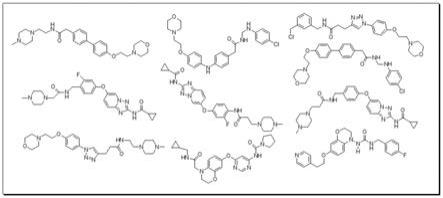
vegfr-2抑制剂的筛选方法、vegfr-2抑制剂和抗肿瘤药物
技术领域
[0001]
本发明涉及药物筛选技术领域,具体而言,涉及vegfr-2抑制剂的筛选方法、vegfr-2抑制剂和抗肿瘤药物。
背景技术:
[0002]
恶性肿瘤是严重威胁人类健康的重大高发疾病之一,我国科学和技术发展规划纲要已将恶性肿瘤的基础和应用研究列为公共卫生与重要疾病防治的关键科学问题。因此,新型抗肿瘤药物的研究不仅是临床肿瘤治疗和提高我国人民健康水平的迫使需要,而且对于我国医药产业和社会发展具有重要意义。
[0003]
肿瘤的生长和转移依赖其周围新生血管的形成,血管内皮生长因子(vascular endothelial growth factor,vegf)是一类由多种组织细胞分泌的高生物活性的功能性糖蛋白,是目前发现的作用最强、特异性最高的促血管生成因子。vegf在脉管生成、血管形成和血管迁移过程中起重要调节作用,并在多种恶性肿瘤中过度表达,与肿瘤的生长、转移、预后关系密切。以抑制血管内皮生长因子受体(vascular endothelial growth factor receptor,vegfr)为基础的抗血管生成疗法已成为癌症治疗中最有效的手段之一。在众多vegf亚型中,vegfr-2是一种特异性糖蛋白,相对分子质量为210000~230000,主要分布在血管内皮细胞和造血干细胞中,可以与vegf-a、vegf-c、vegf-d和vegf-e结合,主要调节vegf在血管内皮细胞中的生理反应,包括通透性、增殖和迁移,是生理性和病理性血管生成过程中的一个关键信号传感器。因此,以抑制血管内皮生长因子受体(vegfr)为基础的抗血管生成疗法已成为癌症治疗中最有效的手段之一。
[0004]
但目前市面已知的vegfr-2抑制剂种类较少,仍需要筛选和研发出更多的vegfr-2抑制剂。
[0005]
鉴于此,特提出本发明。
技术实现要素:
[0006]
本发明的目的在于提供一种vegfr-2抑制剂的筛选方法、vegfr-2抑制剂和抗肿瘤药物。本发明提供的筛选方法是一种全新的筛选方法,能够筛选出新的对vegfr-2具有抑制活性的化合物。
[0007]
本发明是这样实现的:
[0008]
vegfr-2的活性口袋主要由疏水腔组成,有四个亚区与配体结合(参考图1):i区是与铰链区相邻的平坦疏水腔,含有两个重要的氢键受体位点:glu915的c=o作为氢键受体,cys917的nh作为氢键供体。ii区是疏水性侧链(ile886,leu887,ile890,val896,val897,leu1017,ile1042),其由较大的疏水腔构成。-区和-区之间的asp1044骨架上存在由lys866、glu883侧链和羰基氧形成的重要极性区域。在asp1044侧链中的nh可用作键-键供体位点,glu883中的c=o可用作氢键受体。iii区是一个小的疏水区域。iv区是由疏水性leu1033,cys1043和极性氨基酸asn921,arg1030,asn1031组成的极性区域。小分子化合物
通过结合vegfr-2蛋白的这些活性位点(i区-iv区)发挥抑制作用。
[0009]
结合vegfr-2活性腔研究,并结合文献报道的各种小分子抑制剂(如图2所示)和vegfr-2结合模式,以及一些众所周知的抑制剂的一些分子对接结果,其与vegfr-2的结合通常由四个部分组成。以图3的化合物索拉非尼为例:第一部分通常是平坦的疏水基团,第二部分是连接第一部分和第三部分的极性连接链;第三部分是疏水基团(苯环或取代苯环较多);第四部分主要由疏水基团组成,但通常具有氢键受体(glu917)和蛋白质氢键。
[0010]
据此,本发明提出一种全新的筛选vegfr-2抑制剂的构思:即基于vegfr-2的结合活性部位以及现有已知的vegfr-2小分子抑制剂与vegfr-2结合的结合结构特点,根据亲水亲油以及与配体结合部位的不同,将现有已知的vegfr-2小分子抑制剂进行拆分,得到具有结合不同靶点部位的片段,每一个化合物可以拆分成四个不同的片段(一个片段结合一个vegfr-2的结合活性部位),不同化合物拆分出的结合相同活性部位的片段作为一个库,共得到四个片段库;再将来自四个片段库的片段随机组合,形成虚拟化合物库;再使用软件对其进行筛选,获得对vegfr-2具有预测抑制活性的虚拟化合物;再通过体外或体内实验进行验证,进而可以得到一种全新结构不同于现有已知vegfr-2抑制剂的新的vegfr-2抑制剂。
[0011]
基于此,一方面,本发明提供一种vegfr-2抑制剂的筛选方法,其包括:构建虚拟化合物库步骤;
[0012]
所述构建虚拟化合物库步骤包括:
[0013]
拆分步骤:选取多个具有vegfr-2抑制活性的原始化合物作为原始化合物库;将原始化合物库中的每个原始化合物拆分为四个不同的片段,每个原始化合物具有vegfr-2抑制活性,该四个不同的片段能分别结合vegfr-2的四个不同的结合活性部位;该四个不同的结合活性部位分别是第一结合活性部位、第二结合活性部位、第三结合活性部位和第四结合活性部位;将从全部原始化合物拆分出的能够结合所述第一结合活性部位的片段作为第一片段库,能够结合所述第二结合活性部位的片段作为第二片段库,能够结合所述第三结合活性部位的片段作为第三片段库,能够结合所述第四结合活性部位的片段作为第四片段库;
[0014]
组合步骤:从所述第一片段库、所述第二片段库、所述第三片段库和所述第四片段库中各随机选取一个片段构建虚拟化合物,并得到第一虚拟化合物库;
[0015]
类药筛选步骤:根据类药五原则对所述第一虚拟化合物库的化合物进行类药性分析筛选,得到第二虚拟化合物库。
[0016]
需要说明的是,根据现有技术的内容,本领域技术人员能够容易地理解到vegfr-2的四个不同的结合活性部位的结构和位置,即分别对应图1中的i区、ii区、iii区和iv区。
[0017]
需要说明的是,上述的原始化合物现有技术报道的对vegfr-2具有抑制活性的化合物。本领域技术人员根据现有技术的相关数据库、文献等可以很容易地获得这些对vegfr-2具有抑制活性的化合物。原始化合物库即由这些对vegfr-2具有抑制活性的化合物所组成。根据本发明的构思,原始化合物库的原始化合物的数量至少为2个;但也应当说明,本领域技术人员可以根据需要选择合适数量的原始化合物用于构建虚拟化合物库。
[0018]
需要说明的是,每一个原始化合物在拆分时,该原始化合物对应结合不同vegfr-2的结合活性部位的两个主要基团之间存在一些连接基团,这些连接基团可以任意的归属到其中的一个主要基团,例如拆分过程中,也可以将极性基团归属铰链区,非极性基团归属疏
水区,也可以由本领域技术人员根据其常识进行划分,无论如何划分,只要保证拆分出的片段具有结合到vegfr-2的结合活性部位的主要基团即可。
[0019]
类药五原则是本领域常见的药物筛选原则,在构建第一虚拟化合物库后,通过类药五原则一方面可以缩小虚拟化合物库的化合物数量,另一方面可以使得保留的化合物更具有成药性。
[0020]
本发明提供了一种全新的筛选方法,该筛选方法通过对现有抑制分子的拆分和重新组合,可以快递地筛选出对vegfr-2具有抑制活性的新的化合物,实现“无到有”的筛选和发现,丰富了现有的筛选技术,也为以vegfr-2为靶点的药物开发提供了更多的候选vegfr-2抑制剂分子。
[0021]
在可选的实施方式中,所述筛选方法还包括:虚拟筛选步骤;
[0022]
所述虚拟筛选步骤包括:利用药物筛选软件对所述第二虚拟化合物库的化合物进行筛选,获得对vegfr-2具有预测抑制活性但不属于所述原始化合物库的待验证化合物。
[0023]
在可选的实施方式中,利用药物筛选软件对所述第二虚拟化合物库的化合物进行筛选的步骤包括:
[0024]
利用半柔性分子对接工具将所述第二虚拟化合物库的化合物与vegfr-2蛋白进行对接,根据打分高低的顺序,选取打分最高的前x个虚拟化合物,该x个虚拟化合物至少包括1个不属于所述原始化合物库的虚拟化合物;接着对该x个虚拟化合物进行类药性虚拟评价以及聚类分析,获得对vegfr-2具有预测抑制活性的但不属于所述原始化合物库的待验证虚拟化合物。
[0025]
需要说明的是,x的取值是大于0的自然数(例如x=1,10,50,100或150等),其是本领域技术人员可以实际打分高低排序后结果来进行选择的,考虑到打分结果较高的化合物可能包括了原始化合物,因此,所选择的x个虚拟化合物至少包括1个不属于原始化合物库的虚拟化合物。当然,具体的上限数量也是本领域技术人员根据需要进行选择的,本发明对此上限值不作限定。
[0026]
其中,类药性虚拟评价的指标包括蛋白限于血脑屏障穿透性、被动的肠内吸收性、肝毒性、人细胞色素p450、血浆蛋白结合酶结合等,本领域技术人员可以根据需要选择合适的指标对其进行评价。
[0027]
在可选的实施方式中,所述半柔性分子对接工具为ligandfit;采用discovery studio2.1软件的adme模块进行类药性虚拟评价。
[0028]
本发明的实施例研究显示,不同的对接方法具有不同的rmsd值,经研究显示,ligandfit对接其rmsd值最小,结果更为准确。
[0029]
例如,选用索拉非尼与vegfr-2受体晶体结构(pdb:4asd)为研究对象,提取晶体中vegfr-2蛋白为对接蛋白,索拉非尼为配体分子进行libdock、ligandfit和cdocker的对接研究,计算最优构象分子与原始分子的rmsd值。各方法所得最优rmsd结果见表1,可见ligandfit为最优筛选方法。
[0030]
在可选的实施方式中,所述筛选方法还包括:实验验证步骤:
[0031]
将得到的所述待验证虚拟化合物进行体外实验和/或体内实验验证其对vegfr-2的抑制活性;其中,具有抑制活性的待验证虚拟化合物即可作为vegfr-2的抑制剂。
[0032]
需要说明的是,进行实验验证的待验证虚拟化合物的数量是本领域技术人员根据
时间需要进行选择的,可以是一个,也可以是多个,本发明对进入实验验证的待验证虚拟化合物不作限定,无论何种数量都属于本发明的保护范围。
[0033]
需要说明的是,待验证虚拟化合物通过逆合成分析和系统的文献调研以及本领域技术人员的合成经验和现有的实验室条件,待验证虚拟化合物的合成基本没有不可逾越的障碍,本领域技术人员是可以容易合成的。
[0034]
在可选的实施方式中,在所述拆分步骤中,所述原始化合物库中的原始化合物的数量至少为2个。
[0035]
在可选的实施方式中,在所述组合步骤中,利用enumerate library from ligands构建所述第一虚拟化合物库。
[0036]
enumerate library from ligands可通过排列组合的方式把不同片段分子组合在一起。
[0037]
需要说明的是,本领域技术人员容易想到在本发明筛选得到的vegfr-2抑制剂的基础上进一步改造和修饰,以提供更好的抑制活性。因此,以本发明筛选得到的vegfr-2抑制剂为基础结果,进行进一步改造或修饰等所得到的衍生抑制剂也是属于本发明的保护范围。
[0038]
另一方面,本发明提供一种vegfr-2抑制剂,其由如上所述的筛选方法筛选得到。
[0039]
在可选的实施方式中,所述抑制剂的结构式如下:
[0040][0041]
另一方面,本发明还提供一种抗肿瘤药物,其含有如上所述的vegfr-2抑制剂。
附图说明
[0042]
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
[0043]
图1为vegfr-2受体的四大结合空腔的结构示意图。
[0044]
图2为部分现有已报道的具有高抑制活性vegfr2小分子抑制剂。
[0045]
图3为索拉非尼与vegfr2受体的结合平面结构示意图。
[0046]
图4为图2中的vegfr2小分子抑制剂拆分后形成的四个小分子片段库。
[0047]
图5为化合物的类药性分析流程。
[0048]
图6为实施例中筛选得到具有较高预测抑制活性的10个代表化合物。
[0049]
图7为代表化合物与vegfr2蛋白晶体的对接研究(上图:平面图;下图:三维图)。
[0050]
图8为联苯类化合物tm的合成线路图。
[0051]
图9-a为联苯类化合物tm的rmsd曲线,说明其在模拟状态下趋于稳定。
[0052]
图9-b为联苯类化合物tm在vegfr2蛋白内的二维结合图。
[0053]
图9-c为联苯类化合物tm与关键氨基酸结合距离,说明其结合趋于稳定。
[0054]
图9-d为vegfr2蛋白的部分关键氨酸残基的最低能量状态。
具体实施方式
[0055]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
[0056]
以下结合实施例对本发明的特征和性能作进一步的详细描述。
[0057]
实施例1
[0058]
本实施例提供的筛选vegfr-2抑制剂的方法如下:
[0059]
(1)构建虚拟化合物库
[0060]
(a)拆分化合物:
[0061]
以图2中所示的原始化合物作为原始化合物库,根据这些化合物与vegfr-2的四个结合活性部位,将每个原始化合物拆分为四个不同的片段。
[0062]
例如以索拉非尼为例:第一部分通常是平坦的疏水基团,第二部分是连接第一部分和第三部分的极性连接链;第三部分是疏水基团(苯环或取代苯环较多);第四部分主要由疏水基团组成,但通常具有氢键受体(glu917)和蛋白质氢键。例如索拉非尼、瑞戈非尼和卡博替尼的拆分示例如下。
[0063][0064]
该四个不同的结合活性部位分别是第一结合活性部位、第二结合活性部位、第三结合活性部位和第四结合活性部位;将从全部原始化合物拆分出的能够结合所述第一结合活性部位的片段作为第一片段库,能够结合所述第二结合活性部位的片段作为第二片段库,能够结合所述第三结合活性部位的片段作为第三片段库,能够结合所述第四结合活性部位的片段作为第四片段库,同一片段库中相同的片段保留一个即可。图2中所示的原始化合物拆分后形成的四个片段库如图4所示,r1代表第一片段库,r2代表第一片段库,r3代表第一片段库,r4代表第一片段库。
[0065]
(b)组合化合物:
[0066]
利用enumerate library from ligands,从所述第一片段库、所述第二片段库、所
述第三片段库和所述第四片段库各随机选取一个片段构建形成虚拟化合物,片段顺序:r1-r2-r3-r4,通过排列组合的方式,得到含有50400个取代芳杂环的第一虚拟化合物库。
[0067]
(c)类药筛选:
[0068]
根据类药五原则对第一虚拟化合物库的化合物进行类药性分析筛选,筛选出符合类药五原则要求的化合物,得到第二虚拟化合物库,含有20904个虚拟化合物;分析流程参考图5。
[0069]
(2)虚拟筛选
[0070]
利用药物筛选软件如discovery studio2.1软件对第二虚拟化合物库的化合物进行筛选,获得对vegfr-2具有预测抑制活性但不属于原始化合物库的待验证化合物;本实施例的具体筛选方法如下:
[0071]
(a)对接方法的确定
[0072]
选用索拉非尼与vegfr-2受体晶体结构(pdb:4asd)为研究对象,提取晶体中其中vegfr-2蛋白为对接蛋白,索拉非尼为配体分子进行libdock、ligandfit和cdocker的对接研究,计算最优构象分子与原始分子的rmsd值,从而确定模型参数,并确定最适虚拟筛选方法。各方法所得最优rmsd(结果见表1),可见ligandfit为最优筛选方法;并且,为排除筛选过程中过多的假阳性,加入已报道的vegfr-2小分子抑制剂到分子库中,以验证该法的准确性。
[0073]
表1三种对接方法所得两类蛋白对接结果rmsd对比
[0074][0075]
(b)选用对接软件的半柔性分子对接模块ligandfit,首先第二虚拟化合物库与4asd蛋白(vegfr-2蛋白)进行对接,选取打分最高的100个小分子进行逐个分析(主要是分析ligandscore和energy的数值)。选取打分最高的100个化合物,其中含有6个所选已知阳性分子,置信度达95%;在此基础上,采用discovery studio2.1/adme模块进行类药性的虚拟评价(包括血脑屏障穿透性、被动的肠内吸收性、肝毒性、人细胞色素p450、血浆蛋白结合酶结合等);再通过聚类分析,获得10类具有最优预测抑制活性的虚拟化合物(各化合物结果见图6),作为待验证虚拟化合物。
[0076]
(c)将得到待验证虚拟化合物与vegfr2重新对接进一步确认其结合方式,结果显示(图7),待验证虚拟化合物的各个片段都能分别结合到vegfr-2蛋白的各结合活性部位。
[0077]
(3)实验验证
[0078]
(a)合成目标化合物
[0079]
本实施例选取10个待验证虚拟化合物中如下化合物进行合成并验证:
[0080][0081]
名称为:n-(2-(4-甲基哌嗪-1-基)乙基)-2-(4'-(2-吗啉代乙氧基)联苯-4-基)乙酰胺;属于联苯类化合物,本文将其简称为tm。
[0082]
合成tm的工艺线路图见图8,邻苯二甲酰亚胺,碳酸钾固体,缓慢滴加入1,2-二溴乙烷,得化合物3。化合物3的乙腈溶液在碳酸钾固体存在下滴加入n-甲基哌嗪的乙腈溶液,回流反应得黄色油状化合物4,继续与80%水合肼回流反应得油状化合物5(4-甲基哌嗪-1-基)乙胺。化合物6和化合物7在碳酸钾/二氯甲烷体系中溶解,室温反应得棕色液体8,继续与碳酸钾,联硼酸频那醇酯和化合物,pd(pph
3
)
2
cl
2
加热至80℃反应3h,得黄色油状液体10。化合物10在5%naoh溶液水解得化合物11,并与化合物5,hbtu在dmf/tea体系下室温反应得白色目标化合物(tm)。
[0083]
具体合成步骤如下:
[0084]
(a1)合成化合物3:2-(2-溴乙基)异二氢吲哚-1,3-二酮:
[0085][0086]
在250ml圆底烧瓶中加入50mldmf,再加入邻苯二甲酰亚胺(10g,68mmol),碳酸钾固体(18.8g,138mmol),缓慢滴加入1,2-二溴乙烷(38.4g,200mmol),当量比为1:2:2.94,室温搅拌3h。tlc中控,反应完毕,加入200ml纯化水,80ml乙酸乙酯萃取两次,合并有机相,干燥,浓缩除去溶剂,得化合物3粗品17g,收率98%。熔点:80-81℃(现有文献熔点:82-83℃)。
1
h nmr(400mhz;cdcl
3
)δ3.65(t,2h),4.15(t,2h),7.70-7.80(m,2h),7.85-7.95(m,2h)。ms(esi):253[m-h]-。
[0087]
(a2)合成化合物4:2-(2-(4-甲基哌嗪-1-基)乙基)异吲哚啉-1,3-二酮:
[0088][0089]
50ml三口烧瓶中加入20ml乙腈后,根据当量比1:1.47:1.1加入化合物3(5g,19.7mmol),搅拌溶清,加入碳酸钾固体(4g,29mmol),往体系中滴加入n-甲基哌嗪(2.17g,21.7mmol)的5ml乙腈溶液,滴毕,在冷凝回流装置下升温回流搅拌20h。tlc中控,反应完毕,过滤,浓缩得黄色油状化合物3g,收率56%。熔点:103-104℃(文献熔点:104-106℃)。ms(esi):273[m]
+
。
[0090]
(a3)合成化合物5:2-(4-甲基哌嗪-1-基)乙胺:
[0091][0092]
在50ml圆底烧瓶中加入15ml乙醇后,根据当量比1:1.6加入化合物4(3g,11mmol),滴加入1.1g 80%水合肼,混合均匀,在冷凝回流装置下升温至回流搅拌3h。tlc中控,反应完毕,降至室温,过滤,滤液浓缩,得油状化合物5共2.32g,收率75%。
1
h-nmr(400mhz;cdcl
3
):1.35(m,2h),2.27(s,3h),2.30-2.65(m,8h),2.77(t,3h,j=4.1hz);ms(esi):143[m]
+
。
[0093]
(a4)合成化合物8:4-(2-(4-溴苯氧基)乙基)吗啉:
[0094][0095]
在250ml三口烧瓶中,根据当量比1:1:2,加入化合物6(17.2g,0.1mol),化合物7
(14.9g,0.1mol),碳酸钾(27.6g,0.2mol),加入120ml dcm溶解,在冷凝回流装置上室温搅拌过夜,tlc点板监控,反应完全后,将反应液倒入100g冰水中,二氯甲烷萃取,收集有机相,减压蒸干溶剂,得棕色液体24.5g,收率86%。ms(esi):285[m-h]-。
[0096]
(a5)合成化合物10:2-(4'-(2-吗啉代乙氧基)联苯-4-基)乙酸甲酯:
[0097][0098]
在100ml三口烧瓶中,根据当量比2.9:1.2:1,加入碳酸钾(5.7g,41mmol),联硼酸频那醇酯(4.3g,17mmol),化合物9(3.2g,14mmol)以及pd(pph
3
)cl
2
(0.5g,0.7mmol),混合均匀后,通入氮气,加入15ml除氧的dmso,在冷凝回流装置上加热至80℃回流搅拌3h,tlc点板监控。反应完全后,将反应液倒入100ml冰水中,二氯甲烷萃取,收集有机相,无水硫酸钠干燥,减压蒸干溶剂,得黄色液体。将所得的黄色液体加入100ml三口烧瓶中,根据当量比1:2,加入化合物8(2.7g,9.5mmol),碳酸钾(2.6g,19mmol)以及pd(pph
3
)cl
2
(0.33g,0.47mmol),混合均匀后通入氮气置换空气,加热至80℃反应过夜。反应完全后,将反应液倒入80ml冰水中,二氯甲烷萃取,收集有机相,无水硫酸钠干燥,减压蒸干溶剂,所得粗品用色谱硅胶柱纯化,得到黄色油状液体2.0g,收率40%。ms(esi):355[m]
+
。
[0099]
(a6)合成化合物11:2-(4'-(2-吗啉代乙氧基)联苯-4-基)乙酸:
[0100][0101]
在100ml三口烧瓶中加入化合物10(2.0g,5.6mmol),10ml甲醇,23.6ml 5%naoh溶液,混合均匀后,在冷凝回流装置上加热至50℃,反应完全后,减压蒸干溶剂后,缓慢的滴加1n盐酸溶液,有白色固体缓慢析出,ph=6时,析出的固体最多,过滤,滤饼在45℃烘干,得白色固体1.4g,收率74%。ms(esi):341[m]
+
。
[0102]
(a7)合成化合物tm:n-(2-(4-甲基哌嗪-1-基)乙基)-2-(4'-(2-吗啉代乙氧基)联苯-4-基)乙酰胺:
[0103][0104]
在50ml三口烧瓶中,根据当量比1:1.1:1.2,加入化合物11(0.34g,1mmol),0.36ml三乙胺,5mldmf,搅拌,再加入化合物5(0.16g,1.1mmol),hbtu(0.45g,1.2mmol),在冷凝回流装置下室温搅拌0.5h,有大量白色固体析出,过滤,滤饼用乙腈洗涤,干燥,得0.3g白色目标化合物tm,收率65%。ms(esi):466[m]
+
。
[0105]
(b)tm的体外活性测试
[0106]
所得化合物针对vegfr2(wt)进行激酶活性测试。
[0107]
vegfr-2激酶活性通过使用抗磷酸酪氨酸抗体与的alphascreen系统(perkinelmer,usa)进行测定。酶反应条件为:50mm trishcl ph 7.5,5mm mncl2,5mm mgcl2,0.01%tween-20和2mm dtt,加入10um atp、0.1ug/ml含生物素化的聚-glutyr(4:1)以及0.1nm的vegfr-2(millipore,uk)。在与atp产生酶催化之前,化合物和酶催化起始孵育在室温(预孵育)5分钟。该反应通过加入100mm edta,10ug/ml的alphascreen蛋白链菌素供体珠的缓冲液(ph 7.4的hepes缓冲液:62.5mm hepes,250mm氯化钠,0.1%bsa)中猝灭。孵育板在黑暗中过夜温育,然后应用酶标仪进行标记阅读。含有底物但无化合物的酶反应孔作为反应对照。含生物素化的聚-glutyr(4:1)和无atp的酶被用作基础对照。使用graphpad prism 5.01(graphpad software,usa)分析小分子50%抑制的浓度时的抑制激酶活性(ic50值)和95%置信区间(95%ci)。使用非线性回归分析拟合量效曲线,以分别为100和0约束曲线的顶部和底部。每次测定重复进行。
[0108]
以瑞戈非尼作为阳性对照,测定tm的抑制活性。表2给出了tm和瑞戈非尼药理酶活性试验结果。结果表明,tm有温和的vegfr-2抑制活性(ic
50
为38.55μmol),表明tm具有一定的潜在研究价值。
[0109]
表2化合物tm的激酶活性测定结果
[0110]
化合物vegfr-2抑制活性瑞戈非尼55.6nmoltm38.55μmol
[0111]
(c)tm的分子动力学模拟
[0112]
为了进一步明确tm小分子与vegfr-2的结合模式,运用discovery studio 2.5中的minimization and dynamics模块对目标分子的分子动力学模拟进行研究。为了研究优选的小分子tm和vegfr-2蛋白结构之间的连接关系,通过分子动力学模拟,探究了每一个氨基酸残基对vegfr-2抑制剂tm的结合亲和力的定量的能量贡献(图9-a到图9-d显示了tm的分子动力学模拟结果)。tm的rmsd值在540ps模拟时间后达到平衡和均匀的波动(图9-a)。在200到540ps轨道内计算蛋白质骨架的rmsd值,其中数据点在0.927829
±
0.11nm之间。在
540ps生产模拟后,vegfr-2抑制剂tm和关键氨基酸lys868等之间的距离趋于一致后(图9-c),表明系统达到了平衡状态。根据md模拟得到的稳定构象,分析了蛋白质和络合物的相互作用。vegfr-2的疏水袋中,vegfr-2抑制剂tm和lys868形成氢键(图9-b)。根据相互作用能分解,lys868对tm的贡献为-184.914kcal/mol(图9-d)。因此,根据tm的结合模式和化学结构,推测,由含氢供体和受体的酰胺支架连接链,可能在vegfr-2抑制剂中具有重要的药理作用。
[0113]
分子动力学模拟的操作方法如下:
[0114]
利用discovery studio 2.5软件中的minimization,dynamics(heating or cooling),dynamics(equilibration)和dynamics(production)模块进行分子动力力学模拟,并利用analyze trajectory模块分析能量数据。首先,点击discovery studio2.5软件minimization选择pdb文件,最大优化步数设置为10000-20000,rms梯度设置为0.05其他默认,进行能量优化。其次,设置dynamics(heating or cooling)模块中的参数,heating steps 10000-20000,advanced heatingg save restart设置为ture,运行程序。然后,在dynamics(heating or cooling)结果基础上,进行dynamics(equilibration)平衡计算,设置参数input typed molecule,equilibration steps 10000-20000,advanced equiliblation ture,equiliblation restart后,运行程序。再次,利用dynamics(production)模块构建每一个运行时间点上的构象文件。设置参数step:10000-20000,apply shake constra:flase,production save rest:ture,production restart file打开dynamics(equilibration)中的ouput的中的rst文件后,运行程序。最后,利用analyze trajectory计算关键氨基酸残基位点与小分子的能量差以及距离的变化,设置合适的参考标准rmsd reference后运行程序即可在结果中查明。最后,利用origin软件进行图形化处理,得到可视结果图。
[0115]
综上,本发明实施例提供了一种新的筛选vegfr-2抑制剂的方法,该方法基于现有已知vegfr-2抑制剂的结合特点,对其进行片段拆分再组合,得到新结构的虚拟化合物,再对这些虚拟化合物进行筛选和验证,进而得到了不同于现有已知vegfr-2抑制剂结构的且对vegfr-2具有抑制活性的新化合物,该筛选方法是一种全新的筛选方法,能够从无到有筛选出vegfr-2抑制剂,极大地丰富了现有的筛选手段,也为以vegfr-2为靶点的抗肿瘤药物研发提供了更为丰富的候选vegfr-2抑制剂。
[0116]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips