
 商标分类
商标分类  商标转让
商标转让 
具有肝病疗效的化合物的制作方法
 2021-02-02 10:02:25|
2021-02-02 10:02:25| 150|
150| 起点商标网
起点商标网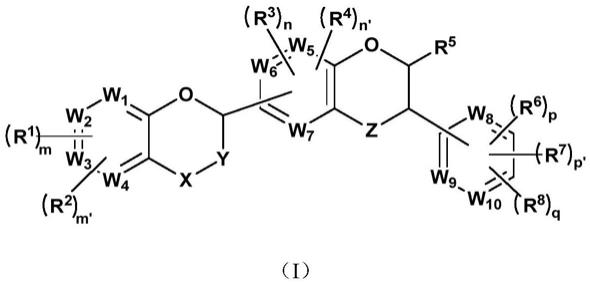
[0001]
本发明属于药物化学领域,具体涉及一类具有肝病疗效的药物化合物及其应用。
背景技术:
[0002]
随着社会发展和生活方式的改变,环境污染、酒精滥用、肥胖症的流行趋势日益加重,脂肪性肝病(fatty liver disease,fld)越来越多的成为终末期肝病发生的重要原因。非酒精性脂肪肝病(non-alcoholic fatty liver disease,nafld)占到fld的70-90%,是一种与遗传、环境及代谢应激相关的临床病理综合征,且逐渐认为是代谢综合征的一部分,其疾病谱主要包括非酒精性单纯性脂肪肝,非酒精性脂肪肝炎(non-alcoholic steatohepatitis,nash)、脂肪性肝纤维化和肝硬化,最终可能演变为肝癌(vlad ratziu,stefano bellentani,helena cortez-pinto,et al.a position statement on nafld/nash based on the easl 2009 special conference.hepatology,2010,53(2):372-384)。nash是单纯性脂肪肝的延续,是指在肝细胞脂肪变性的基础上发生的以肝细胞气球样变、坏死,以及炎性细胞浸润,可伴纤维化为病理特征的慢性肝脏炎症(brent a.neuschwander-tetri,jeanne m.clark,nathan m.bass,et al.clinical,laboratory and histological associations in adults with nonalcoholic fatty liver disease.hepatology,2010,52(3):913-924),报道称大约10-29%的nash患者在未来10年内发展成为了肝硬化(argo ck,caldwell sh.epidemiology and natural history of non-alcoholic steatohepatitis.clinics in liver disease,2009,13(4):511-531)。
[0003]
nafld的患病率不断增高,呈全球化、大众化、低龄化趋势,给人和社会带来极大负担。目前nafld的全球发病率的准确数值尚不确定,因研究人群的不同特征、研究方法和诊断方法等因素的影响,发病率变化很大。中国普通成年人中的发病率约为15-25%(fan jg,farrell gc.epidemiology of non-alcoholic fatty liver disease in china.hepatology,2009,50(1):204-210),西方国家成年人的患病率约为20-30%,其中接近10-25%的nafld演变成nash,而10-15%的nash将发展成肝癌(ong jp,elariny h,collantes r,et al.predictors of nonalcoholic steatohepatitis and advanced fibrosis in morbidly obese patients.obesity surgery,2005,15(3):310-315)。
[0004]
nafld的发病机制尚不明确,目前广为接受的是“二次打击”学说。由脂质代谢紊乱和胰岛素抵抗(ir)所形成的单纯性脂肪肝为“一次打击”,“二次打击”是指“一次打击”后,肝脏大量蓄积脂肪发生氧化应激与脂质过氧化损伤而引起的肝细胞炎症、肝纤维化甚至肝硬化(mesnage r,renney g,seralini ge,et al.multiomics reveal non-alcoholic fatty liver disease in rats following chronic exposure to an ultra-low dose of roundup herbicide.scientific reports,2017(7):39328;娜日苏,包纳日斯.非酒精性脂肪肝的发病机制与流行病学的研究进展.中国医药指南,2016,14(3):39-40)。然而,近年来越来越多的研究显示,nafld的发病与多种因素有关,“二次打击”学说过于简单化,已无法概括nafld的复杂性,因此,“多次击中”理论逐渐取代“二次打击”学说。“多次击中”理
论主要包括胰岛素抵抗、脂肪组织功能障碍、线粒体功能障碍、内质网应激、饮食因素、游离脂肪酸、肠道微生物群失调、慢性炎症状态和遗传因素在nafld发病中的作用。其中,饮食习惯、环境和遗传因素可导致胰岛素抵抗,肥胖与脂肪细胞增殖和肠道微生物组的变化。胰岛素抵抗是脂肪变性发展的关键因素之一,它导致肝脏脂质生成与代谢之间的平衡失调,主要表现为从头脂肪生成增加和脂肪组织脂肪分解减少。进一步,肝脏中的游离脂肪酸增多导致ers和线粒体功能障碍以及随后的炎症反应(fang yl,chen h,wang cl,et al.pathogenesis of non-alcoholic fatty liver disease in children and adolescence:from“two hit theory”to“multiple hit model”.world j gastroenterol,2018,24(27):2974-2983)。
[0005]
nafld的积极防治可阻止慢性肝病进展并改善患者预后。目前尚未证实任何药物对nafld是有益的,所以也没有任何药物获得fda或ema批准。早先人们认为nafld不需要特殊治疗,可以通过饮食调整来防治。目前临床上仍以饮食调整、运动治疗和减肥等为主要措施,但研究报道,饮食调整不能完全有效防止nafld的发展,比如,单纯的降低体重虽可以消除肝脂肪变性,但增加了肝纤维化的程度。因此药物治疗十分关键,nafld尤其是nash的新药研发是近几年兴起的热点。法尼酯x受体激动剂奥贝胆酸是一种合成的亲脂性胆汁酸,2019年2月intercept宣布其在一项iii期临床试验中达到主要终点,显著改善肝脏纤维化。但奥贝胆酸存在明显的剂量相关的瘙痒,超过一半的人在最高剂量时出现瘙痒,9%的人停止了治疗(the farnesoid x receptor(fxr)ligand obeticholic acid in nash treatment trial(flint).nct01265498.2015),在过高剂量下还会导致严重的肝损伤和死亡风险(fda drug safety communication:fda warns about serious liver injury with ocaliva(obeticholic acid)for rare chronic liver disease.september 21,2017)。selonsertib(gs-4997)是细胞凋亡信号调节激酶1(ask1)抑制剂,ii期临床试验中显示出抗纤维化活性,但2019年2月公布的iii期临床试验结果显示其用药48周后未能达到改善肝纤维化的主要目标。genfit的elafibranor是一个pparα/δ双激动剂,可调节脂质平衡和减少肝脏脂肪生成。但在ii期临床试验中,该药未能达到无肝纤维化恶化的脂肪性肝炎消失比例的预设终点,仅对轻中度患者评估达到预期目标(phase iib study to evaluate the efficacy and safety of gft505 versus placebo in patients eithnon-alcoholic steatohepatitis(nash).nct01694849.2012)。因此,目前处于临床期的各类候选药物药效并不显著,且有一定的毒副作用。
[0006]
由于脂质积累在nafld进展中起到关键作用,抑制脂质积累是抗nafld药物开发的主要焦点,长期的临床实践证明,天然药物治疗nafld具有显著的疗效(rodriguez-ramiro i,vauzour d,minihane am.polyphenols and non-alcoholic fatty liver disease:impact and mechanisms.proceedings of the nutrition society,2016,75(1):47-60)。水飞蓟宾是从菊科药用植物水飞蓟种子中提取出来的黄酮类化合物,在治疗肝病方面,水飞蓟宾具有稳定肝细胞膜、保护肝细胞的酶系统和清除肝细胞内活性氧自由基等改善肝功能的作用(rajnarayana k,reddy ms,vidyasaqar j,et al.study on the influence of silymarin pretreatment on metabolism and disposition of metronidazole.arznelmittel-forschung,2004,54:109-113),还具有调节肝脏脂肪代谢的作用,可阻止或改善脂肪在肝脏内的沉积或浸润作用。目前,已有水飞蓟宾用于治疗
nafld的临床实例(王斌,曹燕平,张红旭等.水飞蓟宾治疗非酒精性脂肪性肝炎临床疗效观察,中国肝脏病杂志,2011,3(2):18-21;鲁社玲.水飞蓟宾联合二甲双胍治疗非酒精性脂肪肝.医药论坛杂志,2016,37(4):153-154),但其疗效有限,且水飞蓟宾存在溶解性差和口服生物利用度低等问题,影响其在临床上的疗效。
[0007]
目前已被用于治疗nafld的药物在临床方面存在较多争议,需要更多规范、大样本的临床实验验证其可靠性。临床期药物大多从机制和靶点入手,虽有很多较好的动物实验结果,但在临床上难以重现,或虽已获一些临床实验数据支持,但仍需要更进一步的有效性和安全性研究以支撑其最终上市应用于nafld的治疗。因此,nafld市场亟待疗效佳,毒性低的药物。
技术实现要素:
[0008]
本发明的目的是在现有技术的基础上,提供一类对肝病具有疗效的药物化合物。
[0009]
本发明的另一目的是提供上述化合物在医药方面的用途。
[0010]
本发明的目的可以通过以下措施达到:
[0011]
本发明提供了一类通式(i)所示的化合物、光学异构体或其药学上可接受的盐,
[0012][0013]
其中,
[0014]
r
1
或r
2
分别独立地选自氢、氘、羟基、卤素、氰基、羧基、c
2-4
酯基、c
1-5
烷基、取代的c
1-5
烷基、c
1-5
烷氧基、取代的c
1-5
烷氧基、c
1-3
烷硫基或取代的c
1-3
烷硫基中的一个或多个;
[0015]
m或m
’
分别为0、1或2;
[0016]
x选自o、ch-r
9
、c=o、n-r
9
或共价键;
[0017]
y选自ch-r
10
或c=o;
[0018]
r
3
或r
4
分别独立地选自氢、氘、羟基、卤素、氰基、羧基、c
2-4
酯基、c
1-5
烷基、取代的c
1-5
烷基、c
1-3
烷氧基、取代的c
1-3
烷氧基、c
1-3
烷硫基或取代的c
1-3
烷硫基中的一个或多个;
[0019]
n为0、1或2,n
’
为0或1;
[0020]
z选自o或共价键;
[0021]
r
5
选自氢、氘、羟基、卤素、氰基、氨基、取代氨基、羧基、c
2-4
酯基、c
1-5
烷基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基中的一个或多个;
[0022]
r
6
、r
7
或r
8
分别独立地选自氢、氘、羟基、氨基、取代氨基、硝基、卤素、氰基、羧基、c
2-4
酯基、c
1-5
烷基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基;
[0023]
p或p
’
分别为0或1,q为0、1、2或3;
[0024]
r
9
或r
10
选自氢、氘、羟基、氨基、取代氨基、硝基、卤素、氰基、羧基、c
2-4
酯基、c
1-5
烷
基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基;
[0025]
w
1
、w
2
、w
3
、w
4
、w
5
、w
6
、w
7
、w
8
、w
9
、w
10
分别独立地选自ch或n;
[0026]
r
1
、r
2
、r
3
、r
4
、r
5
、r
6
、r
7
、r
8
、r
9
或r
10
中所述的取代基分别独立地选自氘、羟基、氨基、硝基、卤素、氰基、羧基、c
2-4
酯基、c
1-3
烷基、c
1-3
烷氧基、c
1-3
烷氧-c
1-3
烷氧基、c
1-4
烷氨基或糖基中的一个或多个;
[0027]
且当w
1
、w
2
、w
3
、w
4
、w
5
、w
6
、w
7
同时选自ch时,x不选自ch-r
9
或c=o。
[0028]
在一种优选方案中,通式(i)中,
[0029]
r
1
或r
2
分别独立地选自氢、氘、羟基、卤素、氰基、羧基、c
2-5
酯基、c
1-5
烷基或取代的c
1-5
烷基中的一个或多个;
[0030]
m或m
’
分别为0、1或2;
[0031]
x选自o、ch-r
9
、c=o或共价键;
[0032]
y选自ch-r
10
;
[0033]
r
3
或r
4
分别独立地选自氢、氘、羟基、卤素、氰基、c
1-5
烷基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基中的一个或多个;
[0034]
n为0、1或2,n
’
为0或1;
[0035]
z选自o或共价键;
[0036]
r
5
选自c
1-5
烷基或取代的c
1-5
烷基;
[0037]
r
6
、r
7
或r
8
分别独立地选自氢、氘、羟基、c
1-5
烷基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基;
[0038]
p或p
’
分别为0或1,q为0、1、2或3;
[0039]
r
9
选自氢、氘或羟基;
[0040]
r
10
选自氢、氘、羟基、c
1-5
烷基或取代的c
1-5
烷基;
[0041]
w
1
、w
2
、w
3
、w
4
分别独立地选自ch或n;
[0042]
w
5
、w
6
、w
7
、w
8
、w
9
、w
10
分别选自ch;
[0043]
r
1
、r
2
、r
3
、r
4
、r
5
、r
6
、r
7
、r
8
或r
10
中所述的取代基分别独立地选自氘、羟基、c
1-3
烷氧-c
1-3
烷氧基或卤素中的一个或多个;
[0044]
且当w
1
、w
2
、w
3
、w
4
、w
5
、w
6
、w
7
同时选自ch,x选自ch-r
9
或c=o时,z选自共价键。
[0045]
在一种优选方案中,本发明的化合物选自通式(ii)或(iii)所示的化合物,
[0046][0047]
在一种优选方案中,r
1
或r
2
分别独立地选自氢、羟基、卤素、氰基、羧基、c
2-4
酯基、c
1-3
烷基、取代的c
1-3
烷基、c
1-3
烷氧基、取代的c
1-3
烷氧基、c
1-3
烷硫基或取代的c
1-3
烷硫基中的一个或多个,所述的取代基选自氘、羟基、氨基、硝基、卤素、氰基、羧基、c
2-4
酯基或糖基中的一个或多个。
[0048]
在一种优选的方案中,r
1
或r
2
分别独立地选自羟基、氟、氯、氰基、c
2-4
酯基、c
1-3
烷
基、取代的c
1-3
烷基、c
1-2
烷氧基或取代的c
1-2
烷氧基中的一个或多个,所述的取代基选自氘、羟基、氨基、硝基、氟、氯、氰基或羧基中的一个或多个。
[0049]
在另一种优选的方案中,r
1
或r
2
分别独立地选自氢、氘、羟基、卤素、羧基、c
2-4
酯基、c
1-3
烷基或取代的c
1-3
烷基中的一个或多个,所述的取代基选自氘、羟基、卤素中的一个或多个。
[0050]
在另一种优选的方案中,r
1
或r
2
分别独立地选自氢、羟基、羧基、c
2-4
酯基、c
1-3
烷基、羟基取代的c
1-3
烷基。
[0051]
在另一种优选的方案中,r
1
选自氢、氘、羧基或c
2-5
酯基。
[0052]
在另一种优选的方案中,r
2
选自羟基。
[0053]
在一种优选方案中,m为0、1或2,m
’
为0、1或2。
[0054]
在一种优选方案中,r
3
或r
4
分别独立地选自氢、氘、羟基、卤素、氰基、羧基、c
2-4
酯基、c
1-3
烷基、取代的c
1-3
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基中的一个或多个,其取代基选自氘、羟基、氨基、硝基、卤素、氰基、羧基、c
2-4
酯基或糖基中的一个或多个。
[0055]
在一种优选的方案中,r
3
或r
4
分别独立地选自氢、氘、羟基、卤素、c
1-3
烷氧基或取代的c
1-3
烷氧基中的一个或多个,其取代基选自氘或卤素;
[0056]
在另一种优选的方案中,r
3
或r
4
分别独立地选自氢、氘、羟基、卤素、氰基或c
1-2
烷氧基中的一个或多个。
[0057]
在另一种优选的方案中,r
3
或r
4
分别独立地选自氢、氘、羟基、卤素或c
1-2
烷氧基中的一个或多个。
[0058]
在另一种优选的方案中,r
3
选自氢、氘或c
1-3
烷氧基。
[0059]
在另一种优选的方案中,r
4
选自氢或氘。
[0060]
在一种优选方案中,n为0、1或2,n
’
为0或1。
[0061]
在一种方案中,r
5
选自氢、氘、羟基、卤素、氰基、氨基、c
1-5
烷基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基中的一个或多个,其取代基选自氘、羟基、氨基、硝基、卤素、氰基、羧基、c
2-4
酯基、c
1-3
烷基、c
1-3
烷氧基、c
1-3
烷氧-c
1-3
烷氧基、c
1-4
烷氨基或糖基中的一个或多个。
[0062]
在一种优选的方案中,r
5
优选选自c
1-3
烷基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基,其取代基选自氘、羟基、氨基、硝基、卤素、氰基、c
1-3
烷氧-c
1-3
烷氧基或羧基中的一个或多个。
[0063]
在另一种优选的方案中,r
5
选自c
1-5
烷基、羟基取代的c
1-5
烷基或c
1-3
烷氧-c
1-3
烷氧-c
1-3
烷基。
[0064]
在另一种优选的方案中,r
5
选自羟基取代的c
1-3
烷基或者甲氧甲氧甲基。
[0065]
在一种方案中,r
6
、r
7
或r
8
分别独立地选自氢、氘、羟基、卤素、氨基、氰基、c
1-3
烷基、取代的c
1-3
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基,所述的取代基选自氘、羟基、氨基或卤素中的一个或多个。
[0066]
在一种优选方案中,r
6
或r
7
分别独立地选自羟基、c
1-3
烷氧基或取代的c
1-3
烷氧基,所述的取代基选自氘、羟基、氨基或卤素中的一个或多个。
[0067]
在另一种优选的方案中,r
6
、r
7
或r
8
分别独立地选自氢、羟基、c
1-5
烷基、卤代c
1-5
烷基、c
1-3
烷氧基或卤代c
1-3
烷氧基。
[0068]
在另一种优选的方案中,r
6
、r
7
或r
8
分别独立地选自氢、羟基、c
1-5
烷基或c
1-3
烷氧基。
[0069]
在另一种优选的方案中,r
6
选自c
1-3
烷氧基。
[0070]
在另一种优选的方案中,r
7
选自羟基;r
8
选自氢或氘。
[0071]
在一种更优选方案中,r
8
选自氢、氘、羟基、c
1-3
烷氧基或取代的c
1-3
烷氧基,所述的取代基选自氘、羟基、氨基或卤素中的一个或多个。
[0072]
在一种优选方案中,p或p
’
分别为1,q为0、1、2或3。
[0073]
在一种方案中,x选自o、ch-r
9
、c=o或共价键。
[0074]
在一种方案中,y选自ch-r
10
。
[0075]
在一种优选方案中,r
9
或r
10
分别独立地选自氢、氘、羟基、氨基、硝基、氰基、c
1-3
烷基、取代的c
1-5
烷基、c
1-3
烷氧基或取代的c
1-3
烷氧基,其取代基选自氘、羟基、氨基、硝基、氰基、羧基或糖基中的一个或多个。
[0076]
在一种优选的方案中,r
9
或r
10
优选分别独立地选自氢、氘、羟基、氨基、c
1-3
烷基、c
1-3
羟基烷基或c
1-3
烷氧基。
[0077]
在一种优选的方案中,r
9
选自羟基。
[0078]
在一种优选的方案中,r
10
选自氢、羟基、c
1-3
烷基或羟基取代的c
1-3
烷基。
[0079]
在一种优选的方案中,w
1
、w
2
、w
3
、w
5
、w
6
、w
7
、w
8
、w
9
、w
10
分别选自ch。
[0080]
在一种优选的方案中,w
4
选自ch或n。
[0081]
在另一种方案中,各基团中的r
1
选自氢、氘、羧基或c
2-5
酯基,m为1;r
2
选自羟基,m
’
为1;x选自o或共价键;y选自ch-r
10
,r
10
选自c
1-3
烷基或羟基取代的c
1-3
烷基;r
3
选自氢、氘或c
1-3
烷氧基,n为1;r
4
选自氢或氘,n
’
为0或1;z选自o;r
5
选自羟基取代的c
1-3
烷基;r
6
选自c
1-3
烷氧基,p为1;r
7
选自羟基,p
’
为1;r
8
选自氢或氘,q为0或1;w
1
、w
2
、w
3
、w
4
、w
5
、w
6
、w
7
、w
8
、w
9
、w
10
分别独立地选自ch。
[0082]
在另一种方案中,r
1
选自氢、氘、羧基或甲酸甲酯基,m为1;r
2
选自羟基,m
’
为1;x选自o或共价键;y选自ch-r
10
,r
10
选自甲基或羟甲基;r
3
选自氢、氘或c
1-3
烷氧基,n为1;r
4
选自氢或氘,n
’
为0或1;z选自o;r
5
选自羟甲基;r
6
选自甲氧基,p为1;r
7
选自羟基,p
’
为1;r
8
选自氢或氘,q为0或1;w
1
、w
2
、w
3
、w
4
、w
5
、w
6
、w
7
、w
8
、w
9
、w
10
分别独立地选自ch。
[0083]
在另一种优选的方案中,本发明的化合物选自,
[0084]
[0085][0086]
本发明还涉及一种药物组合物,它以本发明的的化合物、光学异构体或其药学上可接受的盐为活性成分或主要活性成分,辅以药学上可接受的辅料。
[0087]
本发明的化合物、光学异构体或其药学上可接受的盐可以应用在制备治疗或预防肝病的药物方面,特别是应用在制备治疗或预防脂肪肝、肝纤维化或肝硬化药物方面。本发明的化合物、光学异构体或其药学上可接受的盐可以用于治疗或预防肝病的方法,特别是用于治疗或预防脂肪肝、肝纤维化和肝硬化的方法。
[0088]
除非另有说明,下列用在权利要求书和说明书中的术语有如下含义:
[0089]“氢”,是指氕(1h),它是氢元素的主要稳定同位素。
[0090]“氘”,是指氢的一种稳定形态同位素,也被称为重氢,其元素符号为d。
[0091]“羟基”,表示-oh基团。
[0092]“卤素”,是指氟原子,氯原子,溴原子或碘原子。
[0093]“羧基”,表示-cooh基团。
[0094]“烷基”,表示1-20个碳原子的饱和的脂烃基,包括直链和支链基团(本申请书中提到的数字范围,例如“1-20”,是指该基团,此时为烷基,可以含1个碳原子、2个碳原子、3个碳原子等,直至包括20个碳原子)。含1-4个碳原子的烷基称为低级烷基。当低级烷基没有取代基时,称其为未取代的低级烷基。更优选的是,烷基是有2-5个碳原子的中等大小的烷基。本发明中的烷基例如甲基、乙基、丙基、2-丙基、正丁基、异丁基、叔丁基、戊基等。最好是,烷基为有1-4个碳原子的低级烷基,例如甲基、乙基、丙基、2-丙基、正丁基、异丁基或叔丁基等。烷基可以是取代的或未取代的。
[0095]“烷氧基”,表示-o-(未取代的烷基)和-o-(未取代的环烷基)基团,其进一步表示-o-(未取代的烷基)。代表性实施例包括但不限于甲氧基、乙氧基、丙氧基、丁氧基、环丙氧基、环丁氧基、环戊氧基、环己氧基等。
[0096]“c
1-3
烷氧-c
1-3
烷氧基”,表示-o-(未取代的c
1-3
烷基)-o-(未取代的c
1-3
烷基)基团或者-o-(未取代的c
1-3
环烷基)-o-(未取代的c
1-3
环烷基)基团。代表性实施例包括但不限于甲氧甲氧基、甲氧乙氧基、甲氧丙氧基、乙氧甲氧基、甲氧丁氧基、甲氧环丙氧基等。
[0097]“烷硫基”,表示-s-(未取代的烷基)和-s-(未取代的环烷基)基团,其进一步表示-s-(未取代的烷基)。代表性实施例包括但不限于甲硫基、乙硫基、丙硫基、丁硫基、环丙硫基、环丁硫基、环戊硫基、环己硫基等。
[0098]“酯基”,表示2-10个碳原子的羧酸基,包括直链和支链基团,可以用-coo-烷基表示。本发明中的酯基优选采用c
2-5
酯基,进一步优选c
2-4
酯基,更进一步优选c
2-3
酯基;具体的例子例如甲酸甲酯基、甲酸乙酯基、甲酸正丙酯基、甲酸异丙酯基、甲酸丁酯基等。
[0099]“氨基”,表示-nh
2
基团。
[0100]“硝基”,表示-no
2
基团。
[0101]“氰基”,表示-cn基团。
[0102]“共价键”,本发明的共价键特指由两个相邻碳原子通过共用电子并与共用电子之间形成的一种强烈作用的化学键,它还可以称为“碳碳单键”,可以用
“-
(ch
2
)
0
-”
或
“-”
表示。以式(i)为例,当x采用共价键时,y所在环为五元环。
[0103]“羰基”,表示c=o基团。
[0104]“亚甲基”,表示-ch
2-基团。
[0105]“药学上可接受的盐”,是包含通式(i)的化合物与有机酸或无机酸形成的盐,表示保留母体化合物的生物有效性和性质的那些盐。这类盐包括:
[0106]
(1)与酸成盐,通过母体化合物的游离碱与无机酸或有机酸的反应而得,无机酸例如(但不限于)盐酸、氢溴酸、硝酸、磷酸、偏磷酸、硫酸、亚硫酸和高氯酸等,有机酸例如(但不限于)乙酸、丙酸、丙烯酸、草酸、(d)或(l)苹果酸、富马酸、马来酸、羟基苯甲酸、γ-羟基丁酸、甲氧基苯甲酸、邻苯二甲酸、甲磺酸、乙磺酸、萘-1-磺酸、萘-2-磺酸、对甲苯磺酸、水杨酸、酒石酸、柠檬酸、乳酸、扁桃酸、琥珀酸或丙二酸等。
[0107]
(2)存在于母体化合物中的酸性质子被金属离子代替或者与有机碱配位化合所生成的盐,金属离子例如碱金属离子、碱土金属离子或铝离子,有机碱例如乙醇胺、二乙醇胺、三乙醇胺、氨丁三醇、n-甲基葡糖胺等。
[0108]“药物组合物”,指的是在此描述的一种或多种化合物或者它们的药学上可接受的盐和前药与其它的化学成分,例如药学上可接受的载体和赋形剂的混合物。药物组合物的目的是促进化合物对生物体的给药。
[0109]
在下文中,除非特别地限制,作为治疗剂活性成分的式(i)化合物包括它们的所有药学上可接受的盐,它们应当理解为落入本发明的范围内。在本说明书中,仅仅为了方便,将它们简称为“式(i)的化合物”。
[0110]
本发明的化合物、光学异构体或其药学上可接受的盐在肝病方面具有优益的药物活性,可应用于肝病的治疗或预防,特别是在治疗或预防脂肪肝、肝纤维化或肝硬化的药物方面,具有良好的应用前景。
附图说明
[0111]
图1为各受试化合物给药后的斑马鱼油红o染色镜检照片;
[0112]
图中虚线区域所示为肝脏部分:a为正常对照组,b为模型对照组,c为阳性对照物s-腺苷甲硫氨酸组,d为阳性对照物水飞蓟宾组,e为化合物6组,f为化合物14组。
具体实施方式
[0113]
以下结合实施例对本发明做进一步的说明,但本发明的保护范围并不局限于以下各实施例。
[0114]
实施例1:5-羟基-3'-(4-羟基-3-甲氧基苯基)-3,2'-二羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-二{苯并[1,4]二恶烷基}-7-甲酸甲酯(6)的合成
[0115][0116]
步骤a:将含有4-羟基-3,5-二甲氧基苯甲醛(5.0g,27.4mmol)、乙氧甲酰基亚甲基三苯基膦(10.5g,30.2mmol)和二氯甲烷(50ml)的混合物在室温下搅拌过夜。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=1:20洗脱),得3-(4-羟基-3,5-二甲氧基苯基)丙烯酸乙酯(1)(3.11g)。收率为44.9%。
[0117]
步骤b:在-50℃下,向化合物1(3.10g,12.3mmol)的二氯甲烷(30ml)溶液中滴加1.5m二异丁基氢化铝的甲苯溶液(27.0ml)。加完后,在该温度下继续搅拌0.5小时。将反应液缓慢倒入冰水(50ml)中,用2m柠檬酸溶液调节ph值至5~6。用乙酸乙酯(50ml
×
3)萃取,合并的有机相用饱和食盐水(50ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=10:1~4:1洗脱),得4-(3-羟基丙烯基)-2,6-二甲氧基苯酚(2)(2.22g)。收率为86.0%。
[0118]
步骤c:在-50℃下,向3-(4-羟基-3-甲氧基苯基)丙烯酸乙酯(5.0g,22.5mmol)的二氯甲烷(50ml)溶液中滴加1.5m二异丁基氢化铝的甲苯溶液(49.5ml)。加完后,在该温度下继续搅拌0.5小时。将反应液缓慢倒入冰水(50ml)中,用2m柠檬酸溶液调节ph值至5~6。用乙酸乙酯(50ml
×
3)萃取,合并的有机相用饱和食盐水(50ml)洗涤,无水硫酸钠干燥。减
压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=5:1~2:1洗脱),得4-(3-羟基丙烯基)-2,6-二甲氧基苯酚(3)(3.34g)。收率为82.5%。
[0119]
步骤d:将含有没食子酸甲酯(2.03g,11.0mmol)、化合物2(2.32g,11.0mmol)、丙酮(20ml)和苯(40ml)的混合物在60℃搅拌10分钟,然后加入碳酸银(3.03g,11.0mmol)。加完后,所得混合物在该温下搅拌过夜。冷却到室温,过滤除去不溶物。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=1:100~1:60洗脱),得8-羟基-3-(4-羟基-3,5-二甲氧基苯基)-2-羟甲基-2,3-二氢苯并[1,4]二恶烷-6-甲酸甲酯(4)(2.05g)。收率为47.4%。
[0120]
步骤e:在室温下,将三氯化铝(510mg,3.82mmol)分批加入到化合物4(500mg,1.27mmol)的吡啶(5ml)溶液中。加完后,所得混合物在85℃搅拌1小时。冷却到室温,然后加入85%磷酸(1ml),继续搅拌1小时。向混合物中加入水(30ml),用乙酸乙酯(20ml
×
3)萃取,合并的有机相用饱和食盐水(30ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=100:1~40:1洗脱),得3-(3,4-二羟基-5-甲氧基苯基)-8-羟基-2-羟甲基-2,3-二氢-苯并[1,4]二恶烷-6-甲酸甲酯(5)(470mg)。收率为97.5%。
1
h nmr(dmso-d
6
,400mhz)δ9.61(s,1h),8.99(s,1h),8.43(s,1h),7.08(d,j=1.6hz,1h),6.98(d,j=1.6hz,1h),6.56(d,j=1.6hz,1h),6.51(d,,j=1.6hz,1h),4.90(s,1h),4.84(d,j=6.0hz,1h),4.16-4.13(m,1h),3.79(s,3h),3.77(s,3h),3.56-3.54(m,1h),3.43-3.40(m,1h)。
[0121]
步骤f:将含有化合物5(450mg,1.24mmol)、化合物3(226mg,1.24mmol)、丙酮(5ml)和苯(10ml)的混合物在60℃搅拌10分钟,然后加入碳酸银(342mg,1.24mmol),加完后,所得混合物在该温下搅拌过夜。冷却到室温,过滤除去不溶物。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=1:100~1:50洗脱),得5-羟基-3'-(4-羟基-3-甲氧基苯基)-3,2'-双羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-7-甲酸甲酯(6)(398mg)。收率为60.1%。
1
h nmr(dmso-d
6
,400mhz)δ9.63(s,1h),9.13(s,1h),7.08(d,j=1.6hz,1h),7.01-7.00(m,1h),6.99-6.98(m,1h),6.87-6.85(m,1h),6.81-6.80(m,1h),6.73(d,j=1.6hz,1h),6.65(d,j=1.6hz,1h),4.95-4.90(m,4h),4.27-4.22(m,1h),4.15-4.12(m,1h),3.81(s,3h),3.78(s,6h),3.62-3.56(m,2h),3.47-3.42(m,1h),3.41-3.35(m,1h)。ms(esi,m/z):555.1[m-h]-。
[0122]
实施例2:5-羟基-3'-(4-羟基-3-甲氧基苯基)-3,2'-二羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-7-甲酸(7)的合成
[0123][0124]
将含有化合物6(150mg,0.27mmol)、甲醇(5ml)和5m氢氧化钠溶液(5ml)的混合物在40℃下搅拌2小时。冷却到室温,用2m柠檬酸水溶液调节ph值至4~5,加入水(10ml),用乙酸乙酯(15ml
×
3)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=50:1~10:1洗脱),得5-羟
基-3'-(4-羟基-3-甲氧基苯基)-3,2'-二羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-7-甲酸(7)(95mg)。收率为65.1%。
1
h nmr(dmso-d
6
,400mhz)δ12.54(s,1h),9.53(s,1h),9.13(s,1h),7.06(d,j=1.6hz,1h),7.01-7.00(m,1h),6.96-6.95(m,1h),6.87-6.85(m,1h),6.81-6.80(m,1h),6.73(d,j=1.6hz,1h),6.65(d,j=1.6hz,1h),4.95-4.90(m,4h),4.25-4.21(m,1h),4.15-4.12(m,1h),3.81(s,3h),3.79-3.78(m,3h),3.62-3.55(m,2h),3.47-3.39(m,2h)。ms(esi,m/z):541.1[m-h]-。
[0125]
实施例3:3'-(4-羟基-3-甲氧基苯基)-3,7,2'-三羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-5-醇(8)的合成
[0126][0127]
在冰水浴下,向化合物6(150mg,0.27mmol)的thf(5ml)溶液中加入氢化铝锂(41mg,1.08mmol),加完后,所得混合物在室温条件下搅拌5小时。用2m柠檬酸溶液调节ph值至4~5,加入水(20ml),用乙酸乙酯(15ml
×
3)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=60:1~30:1洗脱),得3'-(4-羟基-3-甲氧基苯基)-3,7,2'-三羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-5-醇(8)(50mg)。收率为35.2%。
1
h nmr(dmso-d
6
,400mhz)δ9.13(s,1h),8.99(s,1h),7.01-7.00(m,1h),6.87-6.85(m,1h),6.81-6.79(m,1h),6.70-6.69(m,1h),6.62(d,j=1.6hz,1h),6.40(d,j=1.6hz,1h),6.32(d,j=1.6hz,1h),4.98-4.96(m,1h),4.94-4.89(m,2h),4.87-4.84(m,2h),4.30(s,1h),4.29(s,1h),4.14-4.06(m,2h),3.81(s,3h),3.78-3.77(m,3h),3.61-3.57(m,1h),3.53-3.49(m,1h),3.45-3.41(m,1h),3.38-3.35(m,1h)。ms(esi,m/z):527.1[m-h]-。
[0128]
实施例4:5,8'-二羟基-3'-(4-羟基-3-甲氧基苯基)-3,2'-二羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-7-甲酸甲酯(10)的合成
[0129][0130]
步骤a:在室温下,将三氯化铝(4.11g,30.8mmol)分批加入到化合物4(1.21g,3.08mmol)的吡啶(10ml)溶液中,加完后,在85℃下搅拌过夜。冷却到温室,然后加入85%磷酸(1ml),搅拌1小时。加入水(30ml),用乙酸乙酯(20ml
×
3)萃取,合并的有机相用饱和食盐
水(30ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=100:1~40:1洗脱),得8-羟基-2-羟甲基-3-(3,4,5-三羟基苯基)-2,3-二氢-苯并[1,4]二恶烷-6-甲酸甲酯(9)(490mg)。收率为43.8%。
[0131]
步骤b:将含有化合物9(480mg,1.32mmol)、化合物3(237mg,1.32mmol)、丙酮(5ml)和苯(10ml)的混合物在60℃搅拌10分钟,然后加入碳酸银(437mg,1.58mmol),加完后,所得混合物在该温下搅拌过夜。冷却到室温,过滤除去不溶物。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=1:10~1:50洗脱),得5,8'-二羟基-3'-(4-羟基-3-甲氧基苯基)-3,2'-二羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-7-甲酸甲酯(10)。
1
h nmr(dmso-d
6
,400mhz)δ9.64(s,1h),9.26(s,1h),9.15(s,1h),7.08-7.07(m,1h),7.01-7.00(m,1h),6.97-6.96(m,1h),6.87-6.85(m,1h),6.81-6.79(m,1h),6.53(d,j=1.6hz,1h),6.49(d,j=1.6hz,1h),4.95-4.93(m,1h),4.88-4.83(m,3h),4.17-4.14(m,2h),3.79-3.78(m,6h),3.58-3.56(m,1h),3.51-3.48(m,1h),3.44-3.39(m,2h)。ms(esi,m/z):541.2[m-h]-。
[0132]
实施例5:5,8'-二羟基-3'-(4-羟基-3-甲氧基苯基)-3,2'-二羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-7-甲酸(11)的合成
[0133][0134]
将含有化合物10(47mg,0.087mmol)、甲醇(5ml)和5m氢氧化钠溶液(5ml)的混合物在40℃下搅拌2小时。冷却至室温,用2m柠檬酸水溶液调节ph值至4~5,加入水(10ml),用乙酸乙酯(10ml
×
3)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=1:50~1:30洗脱),得5-羟基-3'-(4-羟基-3-甲氧基苯基)-3,2'-二羟甲基-8'-甲氧基-2,3,2',3'-四氢-[2,6']-联{苯并[1,4]二恶烷基}-7-甲酸(11)。
1
h nmr(dmso-d
6
,400mhz)δ12.47(s,1h),9.54(s,1h),9.26(s,1h),9.15(s,1h),7.06-7.05(m,1h),7.01-7.00(m,1h),6.95-6.94(m,1h),6.86-6.85(m,1h),6.81-6.80(m,1h),6.53(d,j=1.6hz,1h),6.50(d,j=1.6hz,1h),4.94-4.92(m,1h),4.88-4.83(m,3h),4.17-4.11(m,2h),3.79-3.78(m,3h),3.58-3.55(m,1h),3.51-3.48(m,1h),3.44-3.39(m,2h)。ms(esi,m/z):527.2[m-h]-。
[0135]
实施例6:2-[3-(4-羟基-3-甲氧基苯基)-2-羟甲基-2,3-二氢苯并[b][1,4]二恶烷-6-基}-3-甲基-2,3-二氢苯并呋喃-5-酚(14)的合成
[0136][0137]
步骤a:在-10~-5℃下,将三氟化硼乙醚(2.39g,16.8mmol)加入到对苯醌(1.82g,16.8mmol)和异丁香酚甲醚(3.0g,16.8mmol)的二氯甲烷(30ml)溶液中。加完后,所得混合物在该温度下继续搅拌5分钟。加入水(30ml),用乙酸乙酯(30ml
×
3)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:二氯甲烷:石油醚=1:1:15~1:1:7洗脱),得2-(3,4-二甲氧基苯基)-3-甲基-2.3-二氢苯并呋喃)-5-酚(12)(1.0g)。收率为20.8%。
1
h nmr(dmso-d
6
,400mhz)δ8.87(s,1h),7.00-6.94(m,3h),6.62-6.50(m,3h),5.01(d,j=9.2hz,1h),3.75(s,3h),3.74(s,3h),3.32-3.30(m,1h),1.27(d,j=6.8hz,3h)。
[0138]
步骤b:在冰水浴下,将三溴化硼(2.17g,8.66mmol)滴加到化合物12(990mg,3.46mmol)的二氯甲烷(20ml)溶液中,加完后,所得混合物在室温下搅拌3小时。将反应物缓慢倒入到碎冰中(40g),用饱和碳酸氢钠溶液调节ph值至5~6。用乙酸乙酯(30ml
×
3)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=1:10~1:4洗脱),得4-(5-羟基-3-甲基-2,3-二氢苯并呋喃)苯-1,2-二酚(13)(720mg)。收率为80.6%。
1
h nmr(dmso-d
6
,400mhz)δ8.95-8.94(m,3h),6.78-6.50(m,6h),4.91(d,j=8.8hz,1h),3.24-3.20(m,1h),1.26(d,j=6.8hz,3h)。
[0139]
步骤c:将含有化合物13(360mg,1.39mmol)、化合物3(251mg,1.39mmol)、丙酮(8ml)和苯(16ml)的混合物在60℃搅拌10分钟,然后加入碳酸银(500mg,1.81mmol),加完后,所得混合物在该温下搅拌过夜。冷却到室温,过滤除去不溶物。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=1:200~1:80洗脱),得2-[3-(4-羟基-3-甲氧基苯基)-2-羟甲基-2,3-二氢苯并[b][1,4]二恶烷-6-基}-3-甲基-2,3-二氢苯并呋喃-5-酚(14)。
1
h nmr(cdcl
3
,400mhz)δ7.07-6.91(m,6h),6.71-6.59(m,3h),5.06-5.04(m,1h),4.95-4.92(m,1h),4.53(d,j=7.2hz,1h),4.04-4.01(m,1h),3.91(s,3h),3.82-3.77(m,1h),3.59-3.52(m,1h),3.41-3.37(m,1h),1.38(d,j=6.8hz,3h)。ms(esi,m/z):459.2[m+na]
+
。
[0140]
实施例7:3,7-二羟基-2-{8-羟基-3-(4-羟基-3-甲氧基苯基)-2-羟甲基-2,3-二氢苯并[b][1,4]二恶烷-6-基}-2,3-二氢-4h-吡喃并[3,2-b]吡啶-4-酮(25)的合成
[0141][0142]
步骤a:将含有没食子酸甲酯(4.10g,22.3mmol)、化合物3(4.40g,24.4mmol)、氧化银(6.10g,26.3mmol)、丙酮(80ml)和苯(160ml)的混合物在氮气下60℃搅拌10小时。冷却到室温,过滤除去不溶物。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=1:100~1:50洗脱),得8-羟基-3-(4-羟基-3-甲氧基苯基)-2-羟甲基-2,3-二氢苯并[b][1,4]二恶烷-6-羧酸甲酯(15)(3.0g)。收率为37.1%。
[0143]
步骤b:在冰水浴下,将40%氯甲基甲醚(6.20g,30.8mmol)滴加到化合物15(3.0g,8.28mmol)和二异丙基乙胺(6.0g,46.4mmol)的二氯甲烷(60ml)溶液中,加完后,所得混合物在室温下搅拌过夜。加入水(30ml),分层,水层用二氯甲烷(30ml)萃取。合并的有机层用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,石油醚:乙酸乙酯=10:1~3:1洗脱),得3-[3-甲氧基-4-(甲氧基甲氧基)苯基]-8-(甲氧基甲氧基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并[b][1,4]二恶烷-6-羧酸甲酯(16)(2.13g)。收率为52.0%。
[0144]
步骤c:在-10~0℃下,将化合物16(2.10g,4.25mmol)的thf(10ml)溶液滴加到含有氢化铝锂(322mg,8.48mmol)和thf(20ml)的混合物中,加完后,所得混合物在0~5℃下搅拌2小时。反应结束后,向反应混合物中依次缓慢滴入水(0.5ml)、10%氢氧化钠溶液(1.0ml)和水(1.5ml)。通过硅藻土过滤除去不溶物,滤饼用少量乙酸乙酯淋洗。合并的滤液用无水硫酸钠干燥。减压蒸除溶剂,得{3-[3-甲氧基-4-(甲氧基甲氧基)苯基]-8-(甲氧基
甲氧基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并[b][1,4]二恶烷-6-基}甲醇(17)粗品(2.10g)。该化合物不经纯化直接用于下一步反应。
[0145]
步骤d:将含有化合物(17)粗品(2.10g)、二氧化锰(1.90,21.8mmol)和氯仿(30ml)的混合物在43℃搅拌过夜。通过硅藻土过滤除去不溶物,减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,石油醚:乙酸乙酯=6:1~1:3洗脱),得3-[3-甲氧基-4-(甲氧基甲氧基)苯基]-8-(甲氧基甲氧基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并[b][1,4]二恶烷-6-甲醛(18)(1.44g)。步骤c和d两步反应总收率为72.9%。
[0146]
步骤e:在冰水浴下,将2-氰基-3,5-二氯吡啶(10.0g,57.8mmol)的thf(40ml)溶液滴加到含有60%氢化钠(6.90g,173mmol)和thf(40ml)的混合物中,加完后,在该温度下继续搅拌30分钟,然后加入苯甲醇(13.8g,128mmol)。加完后,所得混合物在室温下搅拌过夜。加入饱和食盐水(240ml),用乙酸乙酯(100ml
×
3)萃取,合并的有机相用饱和食盐水(60ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,所得产物用石油醚/二氯甲烷重结晶,得3,5-二苄氧基-2-氰基吡啶(19)(14.0g)。收率为76.6%。
[0147]
步骤f:在-30℃下,将1m甲基溴化镁的thf溶液(121ml)滴加到化合物19(12.8g,40.4mmol)的thf(90ml)溶液中,加完后,所得混合物在该温度下继续搅拌0.5小时,然后在室温下搅拌2小时。用冰水浴冷却后,加入6m盐酸(120ml),然后在该温度下搅拌2小时。用4m氢氧化钠溶液调节ph值至7~8,用乙酸乙酯(100ml
×
3)萃取,合并的有机相用饱和食盐水(60ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,所得产物用石油醚/二氯甲烷重结晶,得1-(3,5-二苄氧基吡啶-2-基)乙-1-酮(20)(12.8g)。收率为95.0%。
[0148]
步骤g:向化合物20(12.7g,38.1mmol)的乙酸乙酯(130ml)溶液中加入5%氢氧化钯(1.30g),所得混合物在氢气中40℃常压下搅拌过夜。通过硅藻土过滤后,减压蒸除溶剂,得1-(3,5-二羟基吡啶-2-基)乙-1-酮(21)(5.40g)。收率为92.6%。
[0149]
步骤h:在冰水浴下,将40%氯甲基甲醚(3.4g,16.9mmol)滴加到化合物21(2.0g,13.1mmol)和二异丙基乙基胺(2.50g,19.3mmol)的二氯甲烷(15ml)溶液中。加完后,在该温度下继续搅拌0.5小时。加入二氯甲烷(45ml),用水(15ml
×
2)洗涤。减压蒸除溶剂,向所得产物中加入由氢氧化钠(1.56g,39.0mmol)和水(8ml)配成的氢氧化钠溶液和四丁基溴化铵(210mg,0.652mmol),然后在冰水浴下滴加40%氯甲基甲醚(2.62g,13.0mmol)。加完后,在该温度下继续搅拌15分钟。加入水(20ml),用二氯甲烷(20ml
×
3)萃取,合并的有机相用饱和食盐水(15ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,石油醚:乙酸乙酯=10:1~5:1洗脱),得1-[3,5-二(甲氧基甲氧基)吡啶-2-基]乙-1-酮(22)(1.90g)。收率为60.1%。
[0150]
步骤i:向化合物18(1.50g,3.23mmol)和化合物22(780mg,3.23mmol)的乙醇(30ml)溶液中加入氢氧化钾(540mg,9.62mmol),加完后,所得混合物在室温下搅拌过夜。加入水(100ml),用二氯甲烷(40ml
×
3)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,石油醚:乙酸乙酯=10:1~1:1洗脱),得1-[3,5-二(甲氧基甲氧基)吡啶-2-基]-3-{3-[3-甲氧基-4-(甲氧基甲氧基)苯基]-8-(甲氧基甲氧基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并[b][1,4]二恶烷-6-基}丙-2-烯-1-酮(23)(1.50g)。收率为67.5%。
[0151]
步骤j:将含有化合物23(1.20g,1.74mmol)、85%间氯过氧苯甲酸(1.55g,
7.63mmol)和乙酸乙酯(20ml)的混合物在40℃搅拌48小时。加入乙酸乙酯(40ml),用饱和碳酸氢钠溶液(20ml
×
2)洗涤,减压蒸除溶剂,得[3,5-二(甲氧基甲氧基)吡啶-2-基]{3-{3-[3-甲氧基-4-(甲氧基甲氧基)苯基]-8-(甲氧基甲氧基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并[b][1,4]二恶烷-6-基}环氧乙烷-2-基}甲酮(24)粗品(1.56g)。该化合物不经纯化直接用于下一步反应。
[0152]
步骤k:向化合物24粗品(1.56g)的甲醇(9ml)和thf(3ml)溶液中滴加浓盐酸(1.2ml),加完后,所得混合物在65℃搅拌1小时。加入水(20ml),用乙酸乙酯(25ml
×
3)萃取,合并的有机相用饱和食盐水(15ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲醇:二氯甲烷=1:50~1:35洗脱),得3,7-二羟基-2-{8-羟基-3-(4-羟基-3-甲氧基苯基)-2-羟甲基-2,3-二氢苯并[b][1,4]二恶烷-6-基}-2,3-二氢-4h-吡喃并[3,2-b]吡啶-4-酮(25)。
1
h nmr(dmso-d
6
,400mhz)δ9.17(s,1h),7.43-7.27(m,4h),7.04-6.94(m,2h),6.81-6.75(m,3h),4.88-4.77(m,2h),4.24-4.17(m,2h),3.77(s,3h),2.85-2.76(m,2h),2.71-2.64(m,2h)。ms(esi,m/z):482.1[m-h]-。
[0153]
实施例8:2-[3-(3,5-二叔丁基-4-甲氧基苯基)-2-羟甲基-2,3-二氢苯并呋喃-5-基]-3,5,7-三羟基色满-4-酮(38)的合成
[0154][0155]
步骤a:将含有2,4,6-三羟基苯乙酮(5.0g,29.7mmol)、氯甲基甲醚(9.7g,120.5mmol)、碳酸钾(37.1g,269mmol)和丙酮(100ml)的混合物在回流下搅拌2小时。减压蒸除溶剂,加入水(50ml),用乙酸乙酯(40ml
×
3)萃取,合并的有机相用饱和食盐水(30ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,甲基叔丁基醚:石油醚=1:15~1:8洗脱),得1-[2-羟基-4,6-二(甲氧基甲氧基)]苯乙酮(26)(5.10g)。收率为67.0%。
1
h nmr(dmso-d6,400mhz)δ13.34(s,1h),6.23(d,j=2.4hz,1h),6.19(d,j=2.4hz,1h),5.30(s,2h),5.23(s,2h),3.44(s,3h),3.38(s,3h),2.60(s,3h)。
[0156]
步骤b:在冰水浴下,将氯甲基甲醚(2.52g,31.3mmol)滴加到含有化合物26(4.0g,15.6mmol)、氢氧化钠(1.84g,46mmol)、水(4ml)、四丁基溴化铵(252mg,0.782mmol)和二氯甲烷(60ml)的混合物中,加完后,所得混合物在室温下搅拌1小时。加入水(40ml),用二氯甲烷(60ml
×
2)萃取,合并的有机相用饱和食盐水(30ml)洗涤,无水硫酸钠干燥。减压蒸除溶
剂,得2,4,6-三(甲氧基甲氧基)苯乙酮(27)(4.60g)。收率为98.2%。
[0157]
步骤c:向5-溴水杨醛(10.0g,49.7mmol)的二氯甲烷(100ml)溶液中加入叔丁基二甲基氯硅烷(9.0g,59.7mmol)和咪唑(4.40g,64.6mmol),加完后,所得混合物在室温下搅拌过夜。加入二氯甲烷(100ml),用水(50ml
×
3)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得5-溴-2-[(叔丁基二甲基硅基)氧]苯甲醛(28)(15.6g)。收率为100%。
[0158]
步骤d:在110℃下,将哌啶(8.4g,98.6mmol)滴加到化合物28(15.6g,49.5mmol)和2,6-二叔丁基苯酚(11.2g,54.3mmol)的甲苯(62ml)溶液中,加完后,所得混合物在装有分水器的装置中回流搅拌过夜。冷却到110℃,然后滴加乙酸酐(10.5g,103mmol),并继续搅拌1小时。冷却到室温,将反应液倒入冰水中,用乙酸乙酯(200ml
×
3)萃取,合并的有机相用饱和食盐水(100ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=1:100~1:50洗脱),得4-{5-溴-2-[(叔丁基二甲基硅基)氧]亚苄基}-2,6-二叔丁基环己-2,5-二烯-1-酮(29)(11.5g)和4-(5-溴-2-羟基亚苄基)-2,6-二叔丁基环己-2,5-二烯-1-酮(30)(3.98g)。收率分别为43.0%和20.7%。
[0159]
步骤e:将含有化合物29(11.5g,21.3mmol)、四丁基氟化铵(6.68g,25.5mmol)和thf(100ml)的混合物在室温下搅拌过夜。加入饱和食盐水(50ml),分层,水层用乙酸乙酯(100ml
×
2)萃取,合并的有机相用饱和食盐水(50ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=1:100~1:50洗脱),得4-(5-溴-2-羟基亚苄基)-2,6-二叔丁基环己-2,5-二烯-1-酮(30)(5.80g)。收率为69.9%。
1
h nmr(dmso-d6,400mhz)δ10.53(s,1h),7.50-7.40(m,4h),7.24(s,1h),6.92(d,j=8.4hz,1h),1.28(s,9h),1.25(s,9h)。
[0160]
步骤f:向化合物30(5.0g,12.8mmol)的乙腈(80ml)溶液中加入(乙氧基羰基甲基)二甲基溴化硫鎓(3.53g,15.4mmol)和碳酸铯(5.02g,15.4mmol),加完后,所得混合物在室温下搅拌2小时。加入水(320ml),用乙酸乙酯(100ml
×
3)萃取,合并的有机相用饱和食盐水(40ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,石油醚洗脱),得5-溴-3-(3,5-二叔丁基-4-羟基苯基)-2,3-二氢苯并呋喃-2-羧酸乙酯(31)(4.50g)。收率为73.9%。
[0161]
步骤g:将含有碳酸钾(870mg,6.30mmol)、碘甲烷(780mg,5.50mmol)、化合物31(2.0g,4.21mmol)和dmf(20ml)的混合物在室温下搅拌过夜。加入水(60ml),用乙酸乙酯(30ml
×
3)萃取,合并的有机相依次用水(15ml
×
2)和饱和食盐水(15ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,石油醚洗脱),得5-溴-3-(3,5-二叔丁基-4-甲氧基苯基)-2,3-二氢苯并呋喃-2-羧酸乙酯(32)(1.60g)。收率为77.6%。
[0162]
步骤h:在冰水浴下,将化合物32(1.60g,3.27mmol)的thf(8ml)溶液滴加到含有氢化铝锂(250mg,6.59mmol)和thf(8ml)的混合物中,加完后,所得混合物在该温度下继续搅拌1小时。然后依次缓慢滴加水(0.25ml)、10%氢氧化钠溶液(0.5ml)和水(0.75ml)。加入乙酸乙酯(40ml),通过硅藻土过滤除去不溶物,滤液用无水硫酸钠干燥。减压蒸除溶剂,得[5-溴-3-(3,5-二叔丁基-4-甲氧基苯基)-2,3-二氢苯并呋喃-2-基]甲醇(33)粗品(1.52g)。该化合物不经纯化直接用于下一步反应。
[0163]
步骤i:在冰水浴下,将氯甲基甲醚(396mg,4.92mmol)滴加到化合物33粗品(1.52g)和二异丙基乙基胺(1.69g,13.1mmol)的二氯甲烷(15ml)溶液中,加完后,所得混合
物在室温下搅拌过夜。加入水(20ml),用二氯甲烷(30ml
×
2)萃取,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,石油醚洗脱),得5-溴-3-(3,5-二叔丁基-4-甲氧基苯基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并呋喃(34)(1.49g)。步骤h和i两步反应总收率为92.7%。
[0164]
步骤j:在-78℃下,将2.5m正丁基锂的正己烷溶液(1.5ml)滴加到化合物34(1.45g,2.95mmol)的thf(15ml)溶液中,加完后,在该温度下继续搅拌1.5小时。然后加入dmf(431mg,6.47mmol),在-78℃下搅拌20分钟后,自然升温到室温搅拌过夜。加入饱和食盐水(30ml),用乙酸乙酯(30ml
×
3)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=1:200~1:50洗脱),得3-(3,5-二叔丁基-4-甲氧基苯基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并呋喃-5-甲醛(35)(490mg)。收率为37.7%。
1
h nmr(dmso-d6,400mhz)δ9.81(s,1h),7.80(d,j=8.0hz,1h),7.53(s,1h),7.10-7.08(m,3h),4.91-4.88(m,1h),4.66-4.58(m,3h),3.84-3.74(m,2h),3.64(s,3h),3.27(s,3h),1.35(s,18h)。
[0165]
步骤k:向化合物27(327mg,1.09mmol)和化合物35(480mg,1.09mmol)的乙醇(10ml)溶液中加入氢氧化钾(183mg,3.26mmol),加完后,所得混合物在室温下搅拌过夜。加入水(30ml),用乙酸乙酯(30ml
×
2)萃取,合并的有机相用饱和食盐水(15ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:石油醚=1:20~1:5洗脱),得3-{3-(3,5-二叔丁基-4-甲氧基苯基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并呋喃-5-基}-1-[2,4,6-三(甲氧基甲氧基)苯基]丙-2-烯-1-酮(36)(554mg)。收率为71.8%。
[0166]
步骤l:将氢氧化钠(151mg,3.78mmol)溶解于水(0.75ml)、二氧六环(4ml)和甲醇(5ml)中,然后依次加入化合物36(544mg,0.753mmol)和30%双氧水(853mg,7.53mmol),加完后,所得混合物在30℃搅拌过夜。加入水(20ml),用乙酸乙酯(20ml
×
2)萃取,合并的有机相用饱和食盐水(20ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得{3-[3-(3,5-二叔丁基-4-甲氧基苯基)-2-[(甲氧基甲氧基)甲基]-2,3-二氢苯并呋喃-5-基]环氧乙烷-2-基}[2,4,6-三(甲氧基甲氧基)苯基]甲酮(37)(541mg)。收率为97.2%。
[0167]
步骤m:向化合物37(531mg,0.719mmol)的甲醇(4ml)和thf(1ml)溶液中滴加浓盐酸(1ml)的甲醇(4ml)溶液,加完后,所得混合物在65℃搅拌1小时。加入水(20ml),用乙酸乙酯(25ml
×
3)萃取,合并的有机相用饱和食盐水(15ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:二氯甲烷:石油醚=1:1:20~1:1:8洗脱洗脱),得2-[3-(3,5-二叔丁基-4-甲氧基苯基)-2-羟甲基-2,3-二氢苯并呋喃-5-基]-3,5,7-三羟基色满-4-酮(38)。
1
h nmr(dmso-d6,400mhz)δ11.90(s,1h),10.85(s,1h),7.34-7.09(m,4h),6.90-6.85(m,1h),5.90-5.80(m,2h),5.11-5.08(m,1h),4.61-4.49(m,3h),3.71-3.63(m,6h),1.36(s,9h),1.35(s,9h)。ms(esi,m/z):563.3[m+h]
+
。实施例9:3
’-
(3,5-二叔丁基-4-甲氧基苯基)-4,6-甲氧基-2
’-
[(甲氧基甲氧基)甲基]-2
’
,3
’-
二氢-[2,5
’-
二苯并呋喃]-3-甲醇(42)的合成
[0168][0169]
步骤a:将在冰水浴下,将氯乙酰氯(9.85g,87.2mmol)滴加到含有间苯三酚(10.0g,79.3mmol)、三氯化铝(21.2g,159mmol)和乙酸乙酯(150ml)的混合物中,加完后,所得混合物在室温下搅拌过夜。将反应混合物缓慢倒入冰水(400ml)中,用乙酸乙酯(150ml
×
3)萃取,合并的有机相用饱和食盐水(80ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,得2-氯-1-(2,4,6-三羟基苯基)乙-1-酮(39)粗品(12.2g)。该化合物不经纯化直接用于下一步反应。
[0170]
步骤b:将含有化合物(39)粗品(12.2g)、碳酸钠(25.0g,236mmol)和乙醇(80ml)的混合物在60℃搅拌过夜。冷却到室温,加入水(250ml),用4m盐酸调节ph值至3~4,用乙酸乙酯(150ml
×
3)萃取,合并的有机相用饱和食盐水(80ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:二氯甲烷:石油醚=1:1:10~1:1:5洗脱),得4,6-二羟基苯并呋喃-3(2h)-酮(40)(4.0g)。步骤a和b两步反应总收率为30.4%。
[0171]
步骤c:将含有化合物(40)(3.0g,18.1mmol)、硫酸二甲酯(5.70g,45.2mmol)、碳酸钾(7.50g,54.3mmol)和乙二醇二甲醚(50ml)的混合物在80℃搅拌过夜。减压蒸除大部分溶剂,加入水(100ml),用乙酸乙酯(50ml
×
3)萃取,合并的有机相用饱和食盐水(30ml)洗涤,无水硫酸钠干燥。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:二氯甲烷:石油醚=1:1:10~1:1:5洗脱),得4,6-二甲氧基苯并呋喃-3(2h)-酮(41)(2.30g)。收率为65.4%。
[0172]
步骤d:将含有化合物34(170mg,0.345mmol)、化合物41(67mg,0.346mmol)、叔丁醇钠(66mg,0.687mmol)、2-双环己基膦-2
’-
甲基联苯(13mg,0.0357mmol)、醋酸钯(4mg,0.0178mmol)和甲苯(10ml)的混合物在60℃搅拌过夜。减压蒸除溶剂,产物经柱层析纯化(200~300目硅胶,乙酸乙酯:二氯甲烷:石油醚=1:1:20~1:1:8洗脱),得3
’-
(3,5-二叔丁基-4-甲氧基苯基)-4,6-甲氧基-2
’-
[(甲氧基甲氧基)甲基]-2
’
,3
’-
二氢-[2,5
’-
二苯并呋喃]-3-甲醇(42)。
1
h nmr(cdcl
3
,400mhz)δ12.61(s,1h),7.75(d,j=8.4hz,1h),7.60(s,1h),7.02(s,2h),6.94(d,j=8.4hz,1h),6.10(d,j=2.0hz,1h),5.82(d,j=2.0hz,1h),4.84-4.80(m,1h),4.75-4.71(m,2h),4.45(d,j=8.4hz,1h),3.91-3.87(m,2h),3.84(s,3h),3.69(s,3h),3.46(s,3h),3.41(s,3h),1.37(s,18h)。ms(esi,m/z):605.2[m+h]
+
。
[0173]
实施例10:化合物对斑马鱼非酒精性脂肪肝的脂肪减少或清除效果试验
[0174]
一、试验材料
[0175]
1.受试化合物
[0176]
受试化合物6和化合物14,分别用dmso配制成40mm的母液备用,-20℃冰箱储存。阳性对照化合物s-腺苷甲硫氨酸(以下简称sam),购自阿拉丁试剂(上海)有限公司,批号为f1523051,用dmso配制成50mm的母液备用。对照化合物水飞蓟宾,购自上海笛柏生物科技有限公司,批号为ee09,用dmso配制成40mm的母液备用。硫代乙酰胺,购自sigma-aldrich,批号为bcbv3031,用dmso配制成1m的母液备用。油红o购自sigma-aldrich,批号slbp5248v。4%多聚甲醛购自鼎国生物科技有限公司,批号773001800。丙二醇购自国药集团化学试剂有限公司,批号20170615。
[0177]
2.试验动物
[0178]
黑色素等位基因突变型半透明albino品系斑马鱼,以自然成对交配繁殖方式进行。鱼龄为受精后3天,每试验组为30尾。
[0179]
以上斑马鱼均饲养于28℃的养鱼用水中(水质:每1l反渗透水中加入200mg速溶海盐,电导作用为480~510μs/cm;ph为6.9~7.2;硬度为53.7~71.6mg/l caco
3
),实验动物使用许可证号为:syxk(浙)2012-0171。饲养管理符合国际aaalac认证的要求。
[0180]
二、试验方法
[0181]
1.斑马鱼非酒精性脂肪肝模型的建立
[0182]
随机选择受精后3天的正常黑色素等位基因突变型半透明albino品系斑马鱼放置于六孔板中,每孔(即每试验组)为30尾,再用终浓度为7mm的硫代乙酰胺处理斑马鱼72小时,即建立斑马鱼非酒精性脂肪肝模型。
[0183]
2.供试化合物的药效评价
[0184]
将斑马鱼转移至六孔板中,随机每孔(即每试验组)30尾。用硫代乙酰胺诱导斑马鱼建立非酒精性脂肪肝模型。将40mm受试化合物6和化合物14定量转入六孔板中,用水稀释至100μm;50mm阳性对照物sam用水配制成终浓度为50μm的剂量组,40mm阳性对照物水飞蓟宾用水配制成终浓度为100μm,同时设置正常对照组(养鱼用水处理斑马鱼)和模型对照组,每孔液体总体积为3ml。除正常对照组外,其余实验组在分别与硫代乙酰胺共同处理72小时后,用油红o进行染色,染色后每个实验组随机选取10尾斑马鱼在解剖显微镜下拍照,用nis-elements d 3.10高级图像处理软件进行图像分析并采集数据,分析统计斑马鱼肝脏脂肪光密度总和(s),各实验组对斑马鱼肝脏脂肪变性抑制作用以以下计算公式,分别评价各个供试品对斑马鱼肝脏脂肪变性抑制率(%),统计学处理结果用mean
±
se表示:
[0185]
肝脏脂肪变性抑制率(%)=[s(模型对照组)-s(供试化合物组)]/[s(模型对照组)-s(正常对照组)]
×
100%
[0186]
用方差分析和dunnett
’
s t-检验进行统计学分析,p<0.05表明具有显著性差异。肝脏脂肪变性抑制率表示受试化合物对造模后的斑马鱼肝脏脂肪的减少程度,数值越大,受试化合物对肝脏脂肪的减少或清除效果越明显。
[0187]
三、试验结果
[0188]
如表1所示,模型对照组斑马鱼肝脏脂肪光密度总和的平均值为22689,显著大于正常对照组的平均值(13098),模型对照组与正常对照组间的统计学分析显示,p值<0.001,表明模型建立成功。经斑马鱼肝脏脂肪光密度总和与模型对照组比较,阳性对照物sam(50μm)对斑马鱼肝脏脂肪变性抑制率为83%(p值<0.001);对照化合物水飞蓟宾(100μm),对斑马鱼肝脏脂肪变性抑制率为45%(p值<0.05)。表明阳性对照物sam和水飞蓟宾均对斑马鱼
非酒精性脂肪肝有保护作用。
[0189]
如表1和图1所示,在100μm下,受试化合物6和化合物14对斑马鱼非酒精性脂肪肝均具有显著的治疗作用,对造模后的斑马鱼肝脏脂肪变性抑制率分别为56%和86%,各组受试化合物斑马鱼肝脏部位的脂滴(油红o染色)明显地减少,而在相同浓度下,阳性对照物水飞蓟宾的抑制率仅为45%,表明这两个化合物对斑马鱼非酒精性脂肪肝的脂肪减少或清除效果远优于水飞蓟宾。
[0190]
表1.供试化合物和水飞蓟宾在100μm浓度下对斑马鱼肝脏脂肪变性抑制的作用(sam浓度为50μm,n=10)
[0191][0192]
与模型对照组比较,*p<0.05,**p<0.01,***p<0.001
[0193]
试验结果表明,本专利的部分化合物对斑马鱼非酒精性脂肪肝具有非常显著的治疗作用,对肝脏脂肪变性抑制率均显著大于水飞蓟宾。
[0194]
各受试化合物给药后的斑马鱼油红o染色镜检照片如图1所示;图中虚线区域所示为肝脏部分:a为正常对照组,b为模型对照组,c为阳性对照物s-腺苷甲硫氨酸组,d为阳性对照物水飞蓟宾组,e为化合物6组,f为化合物14组。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips