
 商标分类
商标分类  商标转让
商标转让 
一种2-苯乙炔硒基醇类化合物的合成方法与流程
 2021-02-02 10:02:56|
2021-02-02 10:02:56| 379|
379| 起点商标网
起点商标网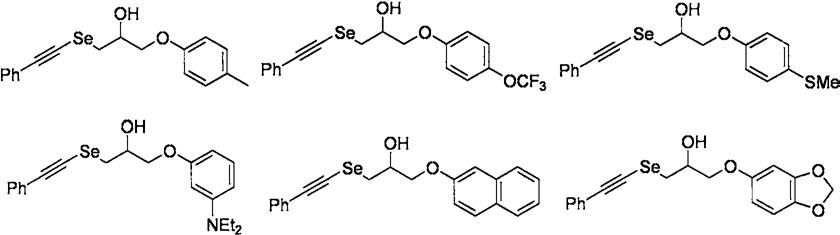
[0001]
本发明属于有机化合物合成技术领域,尤其是涉及一种2-苯乙炔硒基醇类化合物的合成方法。
背景技术:
[0002]
炔基硒醚作为核心骨架广泛存在于具有重要生物活性的天然产物、抗生素、抗氧化药物分子和候选抗癌药物中。此外,炔基硒醚化合物还可以进行双官能团化反应得到多官能化的烯基硒醚化合物,并且通过对其目标产物的后期结构修饰为创新药的发现带来了更多的可能性。因此,探索从简单易得的原料高效地构建炔基硒醚类化合物成为当前有机化学、药物化学和材料科学研究的热点之一。其中,过渡金属铜催化苯丙炔酸的脱羧硒基化反应是构建炔基硒醚化合物的有效方法之一,2017年,brindaban c.ranu课题组(n.mukherjcc,d.kundu,b.c.ranu,adv.synth.catal.,2017,359,329.)报道了铜催化苯丙炔酸与亲电试剂硒氰基苯的脱氰脱羧偶联反应合成炔基硒醚化合物,然而改研究方法所使用的硒氰基苯需要预先制备,并且合成成本昂贵;2018年,南京理工大学的易文斌教授(org.chem.front.,2018,5,428-433)报道了铜催化苯并炔酸与系列硒代布恩特盐的氧化脱羧硒基化反应,然而所合成的硒代布恩特盐及其不稳定,在室温下放置不到一个星期就会变质。此外,2018年,潘英明教授(j.chen,s.-x.su,d.-c.hu,f.-h.cui,y.-l.xu,y.-y.chen,x.-l.ma,y.-m.pan,y.liang,asian j.org.chem.,2018,7,892.)还报道了铜催化苯丙炔酸与二硒醚的脱羧多硒化反应。虽然这些技术为炔基硒醚化合物的合成提供了高效的合成路径,然而这些方法大多存在着原料需要预先制备、使用昂贵的银氧化剂、官能团容忍性差、底物范围窄等诸多缺点。因此,对于简便、易于处理、底物廉价易得的原料来制备炔基烷基硒醚衍生物显得尤为重要,尤其是利用苯丙炔酸、环氧化合物和硒粉的串联反应制备得到2-苯乙炔硒基醇化合物的反应,至今未曾报道,仍存在继续进行研究和探索的必要,这也是本发明得以完成的基础和动力所在。
技术实现要素:
[0003]
本发明所要解决的技术问题是2-苯乙炔硒基醇类化合物的合成路线问题。
[0004]
为解决以上技术问题,本发明提供下述技术方案:
[0005]
一种2-苯乙炔硒基醇类化合物的制备方法,在空气条件下,以苯丙炔酸、硒粉与环氧化合物为反应原料,用水作为反应溶剂,在铜催化剂、配体、相转移催化剂和碱的共同促进作用下,通过串联反应得到2-苯乙炔硒基醇类化合物;
[0006]
上述的反应过程,可用下述的反应式表示:
[0007][0008]
所述苯丙炔酸、环氧化合物与硒粉的摩尔比为1∶3∶3。
[0009]
(1)过渡金属铜催化剂
[0010]
本发明中的过渡金属铜催化剂是醋酸铜、氯化铜、溴化铜或碘化亚铜,优选为氯化铜,以摩尔量计,所述氯化铜的用量与所述苯丙炔酸用量的10%。
[0011]
(2)配体
[0012]
本发明中的配体为三苯基膦、三环己基膦、1,10-邻菲罗啉或2,2
′-
联吡啶,优选为1,10-邻菲罗啉,以摩尔量计,所述配体的用量为所述苯丙炔酸用量的10%。
[0013]
(3)相转移催化剂
[0014]
本发明中的相转移催化剂为氯化四丁基铵、氯化四丁基铵或碘化四丁基铵,优选为碘化四丁基铵,所述相转移催化剂与苯丙炔酸的摩尔比为2∶1。
[0015]
(4)碱
[0016]
本发明中的碱为碳酸铯、碳酸钾、碳酸钠、磷酸钾或磷酸钠中的至少一种,优选碳酸铯,所述碱与苯丙炔酸的摩尔比为3∶1。
[0017]
(6)反应温度
[0018]
本发明的制备方法中,反应温度为30-50℃,非限定性地例如可为30℃、40℃和50℃,反应温度优选50℃。
[0019]
(7)反应时间
[0020]
在本发明的制备方法中,反应时间并无特别的限定,例如可通过液相色谱仪检测目标产物或原料的残留百分比而确定合适的反应时间,其通常为15-24小时,非限定性例如为15小时、18小时、21小时或24小时,反应时间优选24小时。
[0021]
(8)分离纯化
[0022]
在一种优选的实施方式中,反应结束后的后处理步骤可为如下方法:反应结束后,将反应液冷却后加入乙酸乙酯萃取,将有机相用无水硫酸钠干燥,过滤至鸡心瓶,然后旋掉溶剂,将浓缩物通过柱色谱分离,以石油醚和乙酸乙酯混合液为洗脱剂,收集洗脱液,浓缩后得到目标产物。
[0023]
本发明提供的2-苯乙炔硒基醇类化合物的制备方法具有如下有益效果:
[0024]
a)反应高效率、高收率、后处理简便;
[0025]
b)利用廉价易得的硒粉作为硒基化试剂;
[0026]
c)利用水作为绿色反应溶剂;
[0027]
本发明以苯丙炔酸、硒粉与环氧化合物为反应原料,用水作为反应溶剂,在铜催化剂、配体、相转移催化剂和碱的共同促进作用下,通过串联反应得到2-苯乙炔硒基醇类化合物;本发明反应原料廉价易得、产物的产率和纯度高,为2-苯乙炔硒基醇类化合物的制备开拓了合成路线和方法,为硒粉的双官能团化反应提供了新用途,具有重要的社会意义和经济意义。
具体实施方式
[0028]
下面通过具体的实施例对本发明进行详细说明,但这些例举性实施方式的用途和目的仅用来例举本发明,并非对本发明的实际保护范围构成任何形式的任何限定,更非将本发明的保护范围局限于此。
[0029]
以下实施例所给出的新化合物的数据和纯度均通过核磁共振鉴定。
[0030]
实施例1
[0031]
1-苯乙炔硒基-3-(4-甲基苯氧基)-2-丙醇化合物的合成
[0032][0033]
在室温下,将苯丙炔酸(0.2mmol)、单质硒(0.6mmol)、2-(4-甲基苯氧基)甲基环氧乙烷(0.6mmol)、氯化铜(0.02mmol)、1,10-邻菲罗啉(0.02mmol)、碳酸铯(0.6mmol)、四丁基碘化铵(0.4mmol)和2ml水,在50℃反应温度下搅拌24h。反应结束后,加入乙酸乙酯进行稀释,将稀释后的溶液转移至分液漏斗萃取,分离出水相和有机相,再用乙酸乙酯萃取水相3次,合并有机相,加入5g无水硫酸钠,静止30min,每次用5ml乙酸乙酯洗涤滤饼共3次,然后旋掉溶剂,经柱层析分离得到产物(洗脱剂:石油醚∶乙酸乙酯=20∶1),产物为淡黄色液体,收率92%,产物重量为63mg。
[0034]
所得产物的核磁共振氢谱的数据如下:
[0035]
1
h nmr(500mhz,cdcl
3
):δ7.36-7.34(m,2h),7.30-7.24(m,3h),7.05(d,j=8.40hz,2h),6.81(d,j=8.40hz,2h),4.37-4.35(m,1h),4.15-4.09(m,2h),3.18(dd,j=12.50,5.40hz,1h),3.08(dd,j=12.5,5.40hz,1h),2.76(brs,1h),2.27(s,3h);
[0036]
所得产物的核磁共振碳谱的数据如下:
[0037]
13
c nmr(125mhz,cdcl
3
):δ156.2,131.6,130.6,130.0,128.3,128.2,123.2,114.5,99.6,70.5,69.6,69.5,32.8,20.4;
[0038]
对产物进行高分辨质谱的理论计算和实验结果如下:
[0039]
hrms(esi):calcd for c
18
h
18
o
2
se[m+h]
+
347.0545,found 347.0541。
[0040]
实施例2
[0041]
1-苯乙炔硒基-3-(4-三氟甲氧基苯氧基)-2-丙醇化合物的合成
[0042][0043]
在室温下,将苯丙炔酸(0.2mmol)、单质硒(0.6mmol)、2-(4-三氟甲氧基苯氧基)甲基环氧乙烷(0.6mmol)、氯化铜(0.02mmol)、1,10-邻菲罗啉(0.02mmol)、碳酸铯(0.6mmol)、四丁基碘化铵(0.4mmol)和2ml水,在50℃反应温度下搅拌24h。反应结束后,加入乙酸乙酯进行稀释,将稀释后的溶液转移至分液漏斗萃取,分离出水相和有机相,再用乙酸乙酯萃取水相3次,合并有机相,加入5g无水硫酸钠,静止30min,每次用5ml乙酸乙酯洗涤滤饼共3次,然后旋掉溶剂,经柱层析分离得到产物(洗脱剂:石油醚∶乙酸乙酯=20∶1),产物为淡黄色固体,熔点为48-49℃,收率77%,产物重量为64mg。
[0044]
所得产物的核磁共振氢谱的数据如下:
[0045]
1
h nmr(500mhz,cdcl
3
):δ7.35-7.34(m,5h),7.12-7.11(m,2h),6.91-6.89(m,2h),4.39-4.38(m,1h),4.18-4.12(m,2h),3.18(dd,j=12.50,5.40hz,1h),3.08(dd,j=12.50,5.40hz,1h),2.74-2.73(m,1h);
[0046]
所得产物的核磁共振碳谱的数据如下:
[0047]
13
c nmr(125mhz,cdcl
3
):δ156.8,143.2,131.5,128.4,128.3,123.6,123.0,122.4,121.5,119.5,117.5,115.4,99.7,70.7,69.4,69.2,32.7;
[0048]
所得产物的核磁共振氟谱的数据如下:
[0049]
19
f nmr(470mhz,cdcl
3
):δ-58.4(s,3f);
[0050]
对产物进行高分辨质谱的理论计算和实验结果如下:
[0051]
hrms(esi):calcd for c
18
h
15
f
3
o
3
se[m+h]
+
417.0211,found 417.0216。
[0052]
实施例3
[0053]
1-苯乙炔硒基-3-(4-硫甲基苯氧基)-2-丙醇化合物的合成
[0054][0055]
在室温下,将苯丙炔酸(0.2mmol)、单质硒(0.6mmol)、2-(4-硫甲基苯氧基)甲基环氧乙烷(0.6mmol)、氯化铜(0.02mmol)、1,10-邻菲罗啉(0.02mmol)、碳酸铯(0.6mmol)、四丁基碘化铵(0.4mmol)和2ml水,在50℃反应温度下搅拌24h。反应结束后,加入乙酸乙酯进行稀释,将稀释后的溶液转移至分液漏斗萃取,分离出水相和有机相,再用乙酸乙酯萃取水相3次,合并有机相,加入5g无水硫酸钠,静止30min,每次用5ml乙酸乙酯洗涤滤饼共3次,然后旋掉溶剂,经柱层析分离得到产物(洗脱剂:石油醚∶乙酸乙酯=20∶1),产物为淡黄色固体,熔点为63-64℃,收率56%,产物重量为42mg。
[0056]
所得产物的核磁共振氢谱的数据如下:
[0057]
1
h nmr(500mhz,cdcl
3
):δ7.35-7.34(m,2h),7.29-7.22(m,5h),6.88-6.86(m,2h),4.38-4.37(m,1h),4.16-4.11(m,2h),3.19(dd,j=12.50,5.40hz,4h),3.08(dd,j=12.50,5.40hz,1h),2.70-2.69(m,1h),2.43(s,3h);
[0058]
所得产物的核磁共振碳谱的数据如下:
[0059]
13
c nmr(125mhz,cdcl
3
):δ156.8,131.6,129.9,129.8,128.4,128.3,123.1,115.3,99.6,70.5,69.5,69.3,32.7,17.8;
[0060]
对产物进行高分辨质谱的理论计算和实验结果如下:
[0061]
hrms(esi):calcd for c
18
h
18
o
2
sse[m+h]
+
379.0265,found 379.0246。
[0062]
实施例4
[0063]
1-苯乙炔硒基-3-(3-n,n-二乙基苯氧基)-2-丙醇化合物的合成
[0064][0065]
在室温下,将苯丙炔酸(0.2mmol)、单质硒(0.6mmol)、2-(3-n,n-二乙基苯氧基)甲基环氧乙烷(0.6mmol)、氯化铜(0.02mmol)、1,10-邻菲罗啉(0.02mmol)、碳酸铯(0.6mmol)、四丁基碘化铵(0.4mmol)和2ml水,在50℃反应温度下搅拌24h。反应结束后,加入乙酸乙酯进行稀释,将稀释后的溶液转移至分液漏斗萃取,分离出水相和有机相,丙用乙酸乙酯萃取水相3次,合并有机相,加入5g无水硫酸钠,静止30min,每次用5ml乙酸乙酯洗涤滤饼共3次,然后旋掉溶剂,经柱层析分离得到产物(洗脱剂:石油醚∶乙酸乙酯=20∶1),产物为黄色液体,收率83%,产物重量为67mg。
[0066]
所得产物的核磁共振氢谱的数据如下:
[0067]
1
h nmr(500mhz,cdcl
3
):δ7.38-7.36(m,2h),7.28-7.25(m,3h),7.10-7.07(m,1h),6.33-6.31(m,1h),6.24-6.21(m,2h),4.37-4.36(m,1h),4.17-4.11(m,2h),3.30(dd,j=14.0,7.0hz,4h),3.19(dd,j=12.50,5.40hz,1h),3.09(dd,j=12.50,5.40hz,1h),2.74-2.73(m,1h),1.14(s,6h);
[0068]
所得产物的核磁共振碳谱的数据如下:
[0069]
13
c nmr(125mhz,cdcl
3
):δ159.7,149.3,131.6,130.0,128.3,123.2,105.7,100.8,99.6,98.9,70.1,69.6,69.5,44.4,32.9,12.6;
[0070]
对产物进行高分辨质谱的理论计算和实验结果如下:
[0071]
hrms(esi):calcd for c
21
h
25
no
2
se[m+h]
+
404.1123,found 404.1129。
[0072]
实施例5
[0073]
1-苯乙炔硒基-3-(2-萘氧基)-2-丙醇化合物的合成
[0074][0075]
在室温下,将苯丙炔酸(0.2mmol)、单质硒(0.6mmol)、2-(2-萘氧基)甲基环氧乙烷(0.6mmol)、氯化铜(0.02mmol)、1,10-邻菲罗啉(0.02mmol)、碳酸铯(0.6mmol)、四丁基碘化铵(0.4mmol)和2ml水,在50℃反应温度下搅拌24h。反应结束后,加入乙酸乙酯进行稀释,将稀释后的溶液转移至分液漏斗萃取,分离出水相和有机相,再用乙酸乙酯萃取水相3次,合并有机相,加入5g无水硫酸钠,静止30min,每次用5ml乙酸乙酯洗涤滤饼共3次,然后旋掉溶剂,经柱层析分离得到产物(洗脱剂:石油醚∶乙酸乙酯=20∶1),产物为淡黄色固体,熔点为63-64℃,收率90%,产物重量为69mg。
[0076]
所得产物的核磁共振氢谱的数据如下:
[0077]
1
h nmr(500mhz,cdcl
3
):δ7.76-7.71(m,2h),7.65(d,j=8.18hz,1h),7.43-7.40(m,1h),7.35-7.32(m,3h),7.28-7.20(m,3h),7.17-7.15(m,2h),4.48-4.43(m,1h),4.31-4.25(m,2h),3.23(dd,j=12.50,5.40hz,4h),3.13(dd,j=12.50,5.40hz,1h),2.81(brs,1h);
[0078]
所得产物的核磁共振碳谱的数据如下:
[0079]
13
c nmr(125mhz,cdcl
3
):δ156.3,134.4,131.6,129.6,129.3,128.3,128.2,127.6,126.8,126.5,123.9,123.1,118.6,107.1,99.7,70.4,69.6,69.4,32.8;
[0080]
对产物进行高分辨质谱的理论计算和实验结果如下:
[0081]
hrms(esi):calcd for c
21
h
18
o
2
se[m+h]
+
383.0544,found 383.0537。
[0082]
实施例6
[0083]
1-苯乙炔硒基-3-(1,3-亚甲二氧基-4-苯氧基)-2-丙醇化合物的合成
[0084][0085]
在室温下,将苯丙炔酸(0.2mmol)、单质硒(0.6mmol)、2-(1,3-亚甲二氧基-4-苯氧基)甲基环氧乙烷(0.6mmol)、氯化铜(0.02mmol)、1,10-邻菲罗啉(0.02mmol)、碳酸铯(0.6mmol)、四丁基碘化铵(0.4mmol)和2ml水,在50℃反应温度下搅拌24h。反应结束后,加入乙酸乙酯进行稀释,将稀释后的溶液转移至分液漏斗萃取,分离出水相和有机相,再用乙酸乙酯萃取水相3次,合并有机相,加入5g无水硫酸钠,静上30min,每次用5ml乙酸乙酯洗涤滤饼共3次,然后旋掉溶剂,经柱层析分离得到产物(洗脱剂:石油醚∶乙酸乙酯=20∶1),产物为淡黄色液体,收率78%,产物重量为58mg。
[0086]
所得产物的核磁共振氢谱的数据如下:
[0087]
1
h nmr(500mhz,cdcl
3
):δ7.37-7.35(m,2h),7.29-7.27(m,3h),6.67(d,j=
8.45hz,1h),6.50(d,j=2.45hz,1h),6.34(dd,j=8.45hz,2.45hz,1h),5.90(s,2h),4.36-4.33(m,1h),4.11-4.05(m,2h),3.17(dd,j=12.50,5.40hz,4h),3.07(dd,j=12.50,5.40hz,1h),2.72(d,j=5.15hz,1h);
[0088]
所得产物的核磁共振碳谱的数据如下:
[0089]
13
c nmr(125mhz,cdcl
3
):δ153.8,148.3,142.1,131.6,128.3,128.2,123.1,107.9,105.9,101.2,99.6,98.3,71.3,69.5,69.4,32.8;
[0090]
对产物进行高分辨质谱的理论计算和实验结果如下:
[0091]
hrms(esi):calcd for c
18
h
16
o
4
se[m+h]
+
377.0286,found 377.0280。
[0092]
由上述实施例1-6可看出,当采用本发明的所述方法时,能够以高产率、高纯度得到2-苯乙炔硒基醇类化合物。
[0093]
实施例7-9
[0094]
除将其中的催化剂氯化铜分别替换为如下的铜催化剂外,以与实施1相同的方式而分别实施了实施例7-9,所使用铜催化剂和相应产物的收率如下表1所示。
[0095]
表1
[0096]
编号铜催化剂反应产率(%)实施例7醋酸铜不反应实施例8溴化铜40实施例9碘化亚铜22
[0097]
由上表1可看出,当使用其它铜催化剂时,产物产率均大幅度降低。由此证明了本发明所使用的催化剂氯化铜对该反应具有高效催化性能。
[0098]
实施例10-12
[0099]
除将其中的配体1,10-邻菲罗啉分别替换为如下的配体外,以与实施例1相同的方式而分别实施了实施例10-12,所使用配体和相应产物的收率如下表2所示。
[0100]
表2
[0101][0102][0103]
由上表2可看出,当使用膦配体时,没有任何产物,而使用联吡啶为配体时,目标产物产率下降很大,由此证明了本发明所使用的配体1,10-邻菲罗啉对该反应产率的提高极其关键。
[0104]
实施例13-16
[0105]
除将其中的碱碳酸铯分别替换为如下的无机碱外,以与实施例1相同的方式而分别实施了实施例13-16,所使用碱和相应产物的收率如下表3所示。
[0106]
表3
[0107]
编号碱反应产率(%)实施例13碳酸钠不反应实施例14碳酸钾不反应
实施例15磷酸钾不反应实施例16磷酸钠不反应
[0108]
由上表3可看出,当使用其它碱时,几乎均都不反应,由此证明了碳酸铯是该反应成功的关键因素,且对该反应体系最为有效。
[0109]
实施例17-18
[0110]
除将其中的相转移催化剂四丁基碘化铵分别替换为如下的相转移催化剂外,以与实施例1相同的方式而分别实施了实施例17-18,所使用相转移催化剂和相应产物的收率如下表4所示。
[0111]
表4
[0112]
编号相转移催化剂反应产率(%)实施例17四丁基氯化铵42实施例18四丁基溴化铵60
[0113]
由上表4可看出,当使用其它相转移催化剂化合物时,产物产率均下降。由此证明了本发明所使用的相转移催化剂四丁基碘化铵对于该反应具有高效催化性能。
[0114]
综上所述,由上述所有实施例可明确看出,当采用本发明的方法即使用过渡金属铜催化剂(尤其是氯化铜)、配体(1,10-邻菲罗啉)、碱(尤其是碳酸铯)、相转移催化剂(尤其是四丁基碘化铵)和水作反应溶剂,能够使苯丙炔酸、环氧化合物与硒粉发生串联反应而以高产率和高纯度合成得到2-苯乙炔硒基醇类化合物,为该类化合物的高效快捷合成提供了全新的合成路线。
[0115]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然科研对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips