
 商标分类
商标分类  商标转让
商标转让 
一种抗氧剂330的制备方法与流程
 2021-02-02 10:02:08|
2021-02-02 10:02:08| 322|
322| 起点商标网
起点商标网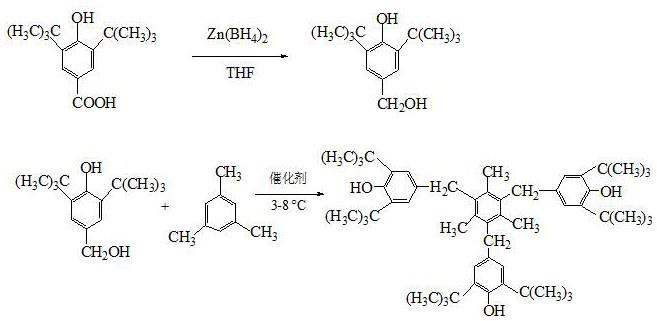
[0001]
本发明涉及化工材料加工技术领域,具体是一种受阻酚抗氧剂330的制备的合成方法,以3,5-二叔丁基-4-羟基苯甲酸为初始原料,经过还原成3,5-二叔丁基-4-羟基苯甲醇,再与均三甲苯在酸性催化剂作用下缩合生成抗氧剂330。
背景技术:
[0002]
抗氧剂330, cas no.:1709-70-2 ,中文名称 1,3,5-三甲基-2,4,6-(3,5-二叔丁基-4-羟基苯甲基)苯 ,是一种高分子量受阻酚类抗氧剂,广泛应用于聚烯烃、pet、pbt、聚酰胺、苯乙烯类树脂及聚氨酯、天然橡胶等弹性体材料。该抗氧剂于1960年由美国壳牌公司开发并推广,具有与树脂相容性好、耐萃取、低挥发、抗氧效率高和电绝缘性好等特点,特别适用于高温加工的聚烯烃管材、注塑制品、电线电缆等制品的加工领域,与亚磷酸酯、硫代酯等辅助抗氧剂和碳自由基捕获剂具有良好的协同效果。
[0003]
正是由于该抗氧剂330的如此优异性质,人们对其合成路线进行了大量的研究。目前为止,有关抗氧剂330的制备工艺、合成方法已经有许多见诸报道,这些报道大分为两类:第一类以2, 6-二叔丁基苯酚和甲醛(或多聚甲醛),在催化剂作用下生产2, 6-二叔丁基-4-羟基卞醇(或2, 6-二叔丁基-4-羟基卞甲醚),再和均三苯在酸性催化剂的条件下,制备出抗氧剂330,这类的研究多集中在2, 6-二叔丁基-4-羟基卞醇(或2, 6-二叔丁基-4-羟基卞甲醚)制备过程催化剂的选择;后一步和均三苯缩合反应过程中酸性催化剂的选择。第二类以4,6-三卤甲基-1,3,5-三甲基苯(卤可为氯、溴)和2, 6-二叔丁基苯酚为原料,在酸性催化剂作用下制备出出抗氧剂330。
[0004]
尽管广大研究人员已经开发了多种受阻酚抗氧剂330的合成方法,但这些方法仍然存在着诸多缺陷,例如反应收率有待提高、反应工艺有待简化、催化剂种类有待开拓等诸多问题。有鉴于此,本发明人旨在通过现有文献的调研与实验探索而开发出一种受阻酚抗氧剂330的新型合成工艺,从而可有效地克服现有技术存在的问题,进而满足高分子材料领域对抗氧剂的广泛需求,具有良好的工业化应用潜力和推广价值。
技术实现要素:
[0005]
本发明人提出了1、一种抗氧剂330的制备方法,其特征在于:以3,5-二叔丁基-4-羟基苯甲酸为初始原料,经过还原制备3,5-二叔丁基-4-羟基苯甲醇,再与均三甲苯缩合生成抗氧剂330,抗氧剂330化学名为1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯。
[0006]
所述以3,5-二叔丁基-4-羟基苯甲酸为初始原料,经过还原成3,5-二叔丁基-4-羟基苯甲醇的过程中,以四氢呋喃thf为溶剂,zn(bh
4
)
2
为还原剂,还原反应温度60-66℃,反应时间5-10小时。
[0007]
所述3,5-二叔丁基-4-羟基苯甲醇与均三甲苯缩合生成抗氧剂330过程中,以二氯甲烷为溶剂,相转移催化剂和浓硫酸的混合复合剂,相转移催化剂为聚乙二醇溶液,反应温
度3-8℃,反应时间2-4小时。
[0008]
所述3,5-二叔丁基-4-羟基苯甲醇与均三甲苯缩合生成抗氧剂330过程中,相转移催化剂为分子量200-1000的聚乙二醇系列。
[0009]
所述酸性催化为相转移催化剂和浓硫酸的混合复合剂,浓硫酸质量浓度85%;质量比m
浓硫酸
:m
聚乙二醇
=100:3.0。
[0010]
所述酸性催化为相转移催化剂和浓硫酸的混合复合剂, 在反应过程中使用量为均三甲苯的2.5倍,m
复合剂
:m
均三甲苯
=2.5:1.0。
[0011]
所述在玻璃反应器内加入均三甲苯、二氯甲烷,冰水浴3-8℃,滴加溶液,1h滴加完后,继续反应3h,液相检测至原料消失,停止反应;将反应液转移至分液漏斗静置分层,分去下层硫酸相,上层有机相用碳酸氢钠溶液洗至中性,蒸馏回收二氯甲烷,用甲苯重结晶得到的白色晶体为最终产物抗氧剂330。
[0012]
所述以3,5-二叔丁基-4-羟基苯甲酸为初始原料,经过还原成3,5-二叔丁基-4-羟基苯甲醇的过程中,以四氢呋喃thf为溶剂,zn(bh
4
)
2
为还原剂,还原反应温度60-66℃,反应时间5-10小时;蒸馏回收溶剂四氢呋喃thf,反应完毕后降至室温后再向体系加入甲醇;搅拌30分钟,过滤体系中的盐,滤液浓缩冷却后析出产品,过滤得产物3,5-二叔丁基-4-羟基苯甲醇。
[0013]
本发明的有益效果如下:第一个方面,本发明以3,5-二叔丁基-4-羟基苯甲酸为初始原料。3,5-二叔丁基-4-羟基苯甲酸的生产工业化程度比较高,是一种易得而且价格比较经济的原料;利用以四氢呋喃thf为溶剂,以zn(bh
4
)
2
为还原剂还原,该步反应收率较高,最优产量可达95%,得到还原产物纯度较高,在工业化过程中,后处理比较简单。因此,以3,5-二叔丁基-4-羟基苯甲酸为初始原料,通过还原为醇来合成抗氧剂330,是一条工业价值较大的工艺路线。
[0014]
第二个方面,在以硫酸与相转移催化剂复合作为缩合反应过程的催化剂。由于相转移催化剂的加入,很大的改善了浓硫酸的催化效果,并减少了浓硫酸的用量,有较大的工业价值。关于该反应催化剂的研究很多,催化剂可选用无机酸、有机酸、lewis酸等,其中有机酸和反应体系混溶,不能通过静置分层的方式分离出体系,需要大量的碱去中和、水洗等缺点; lewis酸酸存在价格贵、用量大、产率不理想、后处理需要加水水解分离等缺点。
[0015]
硫酸具有价格便宜易得,后处理相对方便的特点,仍为目前工业化首选。但由于浓硫酸在反应体系中不相溶,反应中高速搅拌的情况下,催化效率也不是太理想。聚乙二醇类化合物(polyethylene glycols, pegs)看作是开链的冠醚,目前已广泛应用于有机合成中的固-液、液-液等非均相反应。因此,本发明以聚乙二醇作为相转移催化剂来改善这种不足。
具体实施方式
[0016]
一种抗氧剂330的制备方法,以3,5-二叔丁基-4-羟基苯甲酸为初始原料,经过还原制备3,5-二叔丁基-4-羟基苯甲醇,再与均三甲苯缩合生成抗氧剂330,化学名为1,3,5-三甲基-2,4,6-三(3,5-二叔丁基-4-羟基苄基)苯。具体反应方程式为:
。
[0017]
所述以3,5-二叔丁基-4-羟基苯甲酸为初始原料,经过还原成3,5-二叔丁基-4-羟基苯甲醇的过程中,以四氢呋喃thf为溶剂,zn(bh
4
)
2
为还原剂,还原反应温度60-66℃,反应时间5-10小时。
[0018]
所述3,5-二叔丁基-4-羟基苯甲醇与均三甲苯缩合生成抗氧剂330过程中,以二氯甲烷为溶剂,相转移催化剂和浓硫酸的复合作为混合复合剂,反应温度3-8℃,反应时间2-4小时。
[0019]
所述3,5-二叔丁基-4-羟基苯甲醇与均三甲苯缩合生成抗氧剂330过程中,相转移催化剂为分子量200-1000的聚乙二醇系列。
[0020]
所述酸性催化为相转移催化剂和浓硫酸的混合复合剂,浓硫酸为质量浓度85%硫酸;质量比m
浓硫酸
:m
聚乙二醇
=100:3.0。
[0021]
所述酸性催化为相转移催化剂和浓硫酸的混合复合剂, 在反应过程中使用量为均三甲苯的2.5倍,m
复合剂
:m
均三甲苯
=2.5:1.0。
[0022]
所述在玻璃反应器内加入均三甲苯、二氯甲烷,冰水浴3-8℃,滴加溶液,1h滴加完后,继续反应3h,液相检测至原料消失,停止反应;将反应液转移至分液漏斗静置分层,分去下层硫酸相,上层有机相用碳酸氢钠溶液洗至中性,蒸馏回收二氯甲烷,用甲苯重结晶得到的白色晶体为最终产物抗氧剂330。
[0023]
所述以3,5-二叔丁基-4-羟基苯甲酸为初始原料,经过还原成3,5-二叔丁基-4-羟基苯甲醇的过程中,以四氢呋喃thf为溶剂,zn(bh
4
)
2
为还原剂,还原反应温度60-66℃,反应时间5-10小时;蒸馏回收溶剂四氢呋喃thf,反应完毕后降至室温后再向体系加入甲醇;搅拌30分钟,过滤体系中的盐,滤液浓缩冷却后析出产品,过滤得产物3,5-二叔丁基-4-羟基苯甲醇。
[0024]
整个合成工艺分为两步反应:第一步为还原反应:以3,5-二叔丁基-4-羟基苯甲酸为初始原料,以四氢呋喃thf为溶剂,zn(bh
4
)
2
为还原剂,经过还原制备3,5-二叔丁基-4-羟基苯甲醇;第二步为缩合反应:上步反应合成出的3,5-二叔丁基-4-羟基苯甲醇和均三甲苯在浓硫酸和聚乙二醇(分子量200-1000)的复合催化下,缩合生成抗氧剂330。
[0025]
本发明具体实施操作细节如下:(1)还原反应过程:
ꢀ
在玻璃反应器中,分别加入3,5-二叔丁基-4-羟基苯甲酸、 thf(四氢呋喃)、zn(bh
4
)
2
;缓慢升温至60-66℃,反应时间6-8小时,取样检测,达到反应要求即可停止反应。反应完毕后蒸馏回收溶剂四氢呋喃thf;降至室温后再向体系加入300克甲醇,搅拌30分钟,过滤体系中的盐,滤液浓缩冷却后析出产品,过滤得产物3,5-二叔丁基-4-羟基苯甲醇。
[0026]
(2)缩合反应过程:在玻璃反应器内加入均三甲苯、二氯甲烷,3-8℃冰水浴,开始滴加复合催化剂和3,5-二叔丁基-4-羟基苄基甲基醇的二氯甲烷溶液,1h滴加完后,继续反应3h左右,液相检测至原料消失,停止反应。将反应液转移至分液漏斗静置分层,分去下层硫酸相,上层有机相用碳酸氢钠溶液洗至中性,蒸馏回收二氯甲烷,用甲苯重结晶得到的白色晶体为最终产物抗氧剂330。
[0027]
实施例1(1)还原:分别称取3,5-二叔丁基-4-羟基苯甲酸82.5克(0.33mo1),四氢呋喃200ml,还原剂zn(bh
4
)
2 16克(0.17mo1) 加入玻璃反应器中,搅拌升温至63℃,反应时间7小时,取样检测合格后,在80℃蒸馏回收四氢呋喃;向体系加入300克甲醇萃取产物,滤盐浓缩后得到大量晶体析出,该产物为3,5-二叔丁基-4-羟基苯甲醇,收率95.0%。
[0028]
(2)缩合:将上述反应的到的产物(3,5-二叔丁基-4-羟基苯甲醇)27.2g(0.1088mo1) 溶解于100ml二氯甲烷〕待用;在玻璃反应器内,加入均三甲苯4.3克(0.035mol)、二氯甲烷100ml,冰水浴8℃,开始滴加85%硫酸/聚乙二醇400混合物10.75克;同时滴加3,5-二叔丁基-4-羟基苄基甲基醚的二氯甲烷溶液,1h滴加完后,继续反应3h左右至原料消失,停止反应。将反应液转移至分液漏斗,分去下层水相,上层有机相水洗至中性。蒸馏回收二氯甲烷,用甲苯重结晶得到的白色晶体为最终产物抗氧剂330,收率为85.5%。
[0029]
实施例2(1)还原:分别称取3,5-二叔丁基-4-羟基苯甲酸82.5克(0.33mo1),四氢呋喃200ml,还原剂zn(bh
4
)
2 16克(0.17mo1) 加入玻璃反应器中,搅拌升温至66℃,四氢呋喃开始回流,反应时间5小时,取样检测合格后,在80℃蒸馏回收四氢呋喃;向体系加入300克甲醇萃取产物,滤盐浓缩后得到大量晶体析出,该产物为3,5-二叔丁基-4-羟基苯甲醇,收率92.5%。
[0030]
(2)缩合:将上述反应的到的产物(3,5-二叔丁基-4-羟基苯甲醇)27.2g(0.1088mo1) 溶解于100ml二氯甲烷待用;在玻璃反应器内,加入均三甲苯4.3克(0.035mol)、二氯甲烷100ml,冰水浴3℃,开始滴加85%硫酸/聚乙二醇200混合物10.75克;同时滴加3,5-二叔丁基-4-羟基苄基甲基醚的二氯甲烷溶液,1h滴加完后,继续反应4h左右至原料消失,停止反应。将反应液转移至分液漏斗,分去下层水相,上层有机相水洗至中性。蒸馏回收二氯甲烷,用甲苯重结晶得到的白色晶体为最终产物抗氧剂330,收率为81.5%。
[0031]
实施例3(1)还原:分别称取3,5-二叔丁基-4-羟基苯甲酸82.5克(0.33mo1),四氢呋喃200ml,还原剂zn(bh
4
)
2 16克(0.17mo1) 加入玻璃反应器中,搅拌升温至60℃,反应时间10小时,取样检测合格后,在80℃蒸馏回收四氢呋喃;向体系加入300克甲醇萃取产物,滤盐浓缩后得到大量晶体析出,该产物为3,5-二叔丁基-4-羟基苯甲醇,收率94.2%。
[0032]
(2)缩合:将上述反应的到的产物(3,5-二叔丁基-4-羟基苯甲醇)27.2g(0.1088mo1) 溶解于100ml二氯甲烷待用;在玻璃反应器内,加入均三甲苯4.3克
(0.035mol)、二氯甲烷100ml,冰水浴3℃,开始滴加85%硫酸/聚乙二醇600混合物10.75克;同时滴加3,5-二叔丁基-4-羟基苄基甲基醚的二氯甲烷溶液,1h滴加完后,继续反应4h左右至原料消失,停止反应。将反应液转移至分液漏斗,分去下层水相,上层有机相水洗至中性。蒸馏回收二氯甲烷,用甲苯重结晶得到的白色晶体为最终产物抗氧剂330,收率为84.0%。
[0033]
实施例4(1)还原:分别称取3,5-二叔丁基-4-羟基苯甲酸82.5克(0.33mo1),四氢呋喃200ml,还原剂zn(bh
4
)
2 16克(0.17mo1) 加入玻璃反应器中,搅拌升温至63℃,反应时间7小时,取样检测合格后,在80℃蒸馏回收四氢呋喃;向体系加入300克甲醇萃取产物,滤盐浓缩后得到大量晶体析出,该产物为3,5-二叔丁基-4-羟基苯甲醇,收率93.8%。
[0034]
(2)缩合:将上述反应的到的产物(3,5-二叔丁基-4-羟基苯甲醇)27.2g(0.1088mo1) 溶解于100ml二氯甲烷待用;在玻璃反应器内,加入均三甲苯4.3克(0.035mol)、二氯甲烷100ml,冰水浴8℃,开始滴加85%硫酸/聚乙二醇1000混合物10.75克;同时滴加3,5-二叔丁基-4-羟基苄基甲基醚的二氯甲烷溶液,1h滴加完后,继续反应2h左右至原料消失,停止反应。将反应液转移至分液漏斗,分去下层水相,上层有机相水洗至中性。蒸馏回收二氯甲烷,用甲苯重结晶得到的白色晶体为最终产物抗氧剂330,收率为82.5%。
[0035]
本发明已公开的实施例1至4中最终产物抗氧剂330的总收率大于80%以上,与传统工艺相比收率得到大幅提升;而且很大的改善了催化效果,并减少了无机酸的用量,有较大的工业价值。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips