
 商标分类
商标分类  商标转让
商标转让 
矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的对照品的制备方法与流程
 2021-02-02 10:02:04|
2021-02-02 10:02:04| 381|
381| 起点商标网
起点商标网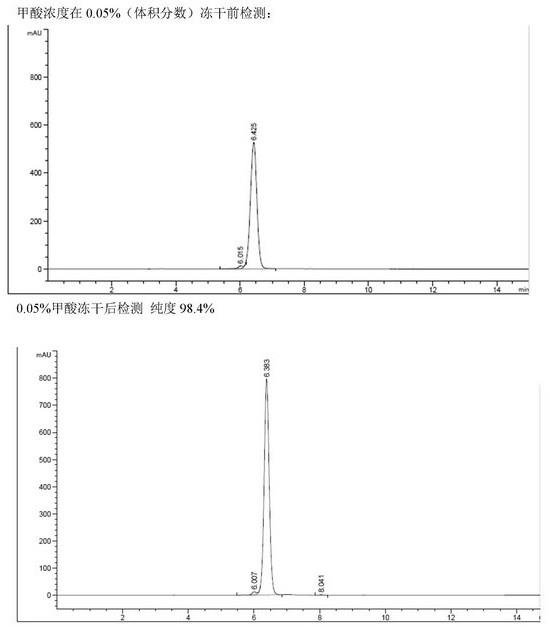
[0001]
本发明涉及一种矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的制备方法,尤其涉及来自紫甘蓝的高纯度的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的固体对照品的制备方法。
背景技术:
[0002]
矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷是一种花色苷,结构如下述的式1所示,其最初被ole-p. johansen等人报道结构(phytochemistry,1991,30, 4137-4141)。
[0003]
式1研究人员还发现其在紫甘蓝中存在。紫甘蓝是常见蔬菜,又称紫洋白菜、红甘蓝、紫椰菜、赤球甘蓝,是结球甘蓝(洋白菜)的变型,属十字花科、芸薹属、甘蓝种,是结球甘蓝的一个变种,最早起源于地中海以及欧洲西部和南部地区,目前在世界各地均有种植、食用,一直是药食同源产品的研究热点。近年来,花色苷的研究成果很多,一般认为,花色苷具有一定的生理活性,如改善视觉功能、改善大脑功能、抗肥胖症、抗糖尿病、抗心血管疾病和抗癌症等。
[0004]
紫甘蓝与普通甘蓝在花色苷含量上区别较大,花色苷和紫甘蓝食疗药用价值关系密切,为了深入研究紫甘蓝的成分含量、营养活性,以及在制定相应的质量标准的探索中,利用花色苷的对照品对紫甘蓝的提取物进行定性和定量检测是最基本的手段。
[0005]
然而,作为紫甘蓝重要成分的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷,目前再市面上尚无其对照品可以购买,使得紫甘蓝的相关研究陷入难以推进的状态。科研界和产业界都迫切需要研究开发一种矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷对照品的制备方法。
[0006]
已经报道的花色苷提取方法有:溶剂提取法、微波和超声波辅助提取法、生物酶或生物发酵法和超高压法等。然而紫甘蓝中的花色苷很多都带有酰基化残基,上述方法在面临紫甘蓝的酰基化花色苷提取时由于分辨率不高,因而很难得到令人满意的效果。本发明
的发明人研究了大量现有文献,发现目前尚不曾有文献提供矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷高纯度样品制备的技术方案。
[0007]
专利文件cn106317145a中,公开了紫甘蓝中cyanidin-3-(sinapoyl) diglucoside-5-glucoside和cyanidin-3
ꢀ-
(caffeoyl) (sinapoyl) diglucoside-5-glucoside两种酰基的花色苷的分离制备方案。但是并未提及矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷,而且其提供的方案仅能获得纯度92.4-94.3%的花色苷,并不能提供纯度达到于98%以上的花色苷。本发明的发明人利用其记载的方案从紫甘蓝中分离制备矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷,发现逆流色谱对于矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的分离效果非常差,完全无法获得高纯度的产物。
[0008]
另外,化合物对照品作为需要分发、出售、存储的试剂,需要以固体形式提供,否则难以维持稳定状态,然而如何能提供一种固体形式且纯度达到98%以上的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的固体对照品的制备方法,是业界迫切需要解决的技术问题。
技术实现要素:
[0009]
本发明的目的在于提供一种纯度达到98%以上的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷(以下,有时也称为目标物)的对照品的制备方法。
[0010]
如果并未特殊说明,本发明中目标物的纯度和含量的百分数,为以质量比计的含量。如果并未特殊说明,本发明中液体与液体含量配比的百分数,例如甲酸在乙腈水溶液中的含量配比,以体积分数计量。
[0011]
本发明通过简单高效的操作流程,能够分离得到纯度很高的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷。本发明的分离方法包括以下步骤:依次包括以下步骤:高压反相色谱柱分离工序,将含有矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的粗品上样至高压反相色谱柱,以5~15.0mpa压力,以含有0.05~1%的甲酸和15%-20%的乙腈的水进行洗脱,获得目标物的溶液,固体化工序,将反相色谱柱制备工序得到目标物的溶液,浓缩至矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的浓度为5%~50%,将浓缩液进行冷冻干燥,得到矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的甲酸盐的固体粉末。
[0012]
作为本发明优选的制备方法,通过依次包含以下工序的方法制备上述在高压反相色谱柱分离工序中作为原料的含有矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的粗品:富集浓缩工序,以甘蓝红提取物为原料,利用酸化的醇类溶剂进行提取,浓缩得到第一原料浓缩液,将第一原料浓缩液上样至苯乙烯类骨架大孔树脂,用水洗脱去除杂质,再利用酸化的醇类溶剂洗脱获得含目标物的溶液,浓缩得到第二原料浓缩液的工序,甘蓝红提取物是利用紫甘蓝蔬菜提取的色素粗品,可以在市场购买,不同厂家批号的产品都可使用。
[0013]
色谱柱提纯工序,将第二原料浓缩液上样至中压反相色谱柱,以压力为0.05~0.5mpa进行梯度洗脱,洗脱过程为,以酸性的4%~5%乙腈的水溶液洗脱去除杂质,再以酸性的7%~9%乙腈的水洗脱收集该洗脱液浓缩得到第一粗品浓缩液;将第一粗品浓缩液上样至羟丙基葡聚糖凝胶色谱柱,利用酸性的含18~22%乙腈的水进行洗脱,利用hplc紫外检测器检测,根据吸收峰形收集样品溶液,浓缩干燥得到含有矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的粗品。
[0014]
作为优选的本发明的制备方法,色谱柱提纯工序中,所述酸性的4%~5%乙腈的水溶液为含有0.03%~0.07%的三氟乙酸的乙腈的水溶液,所述酸性的7%~9%乙腈的水溶液为含有0.03%~0.07%的三氟乙酸的乙腈的水溶液,所述酸性18~22%乙腈的水溶液为含有0.03%~0.07%的三氟乙酸的乙腈的水溶液。
[0015]
作为优选的本发明的制备方法,中压反相色谱柱的填料为粒径为40-60μm的十八烷基硅烷键合硅胶,高压反相色谱柱的填料为粒径为5-10μm的十八烷基硅烷键合硅胶。
[0016]
作为优选的本发明的制备方法,高压反相色谱柱的压力为8.0~10.0 mpa。
[0017]
作为优选的本发明的制备方法,高压反相色谱柱的压力为0.1mpa~0.2mpa。
[0018]
作为优选的本发明的制备方法,对高压反相色谱柱的洗脱,使用0.1~0.2%的甲酸和15%-20%的乙腈的水进行洗脱。
[0019]
作为优选的本发明的制备方法,固体化工序中,将色谱柱制备工序得到目标物的溶液进行浓缩时,在45℃以下的温度减压除去乙腈后,浓缩过程控制在60分钟以内,然后直接进行冷冻干燥。
[0020]
本发明具有以下特点:本发明通过上述方法得到矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的固体粉末纯度可以达到98%以上,是首次提供可行的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的对照品的制备方案,该方法简便易行,而且所有的步骤都适合进行工艺放大,可以进行工业化的制备生产。
[0021]
花色苷一般可以以吡喃阳离子形式与酸阴离子形成类盐形式的固体粉末,本发明的过程中,对矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的稳定性进行了验证,发现其甲酸盐能够稳定存在,从而获得了固体粉末性质的对照品,非常有利于对照品的储藏和分发。还发现目前花色苷类对照品常见的盐酸盐难以稳定存在,难以提供固体粉末纯度可以达到98%以上矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的盐酸盐。
[0022]
附图说明
[0023]
图1为实施例1中固体产品回收工序中目标物冻干前后的hplc图谱;图2为实施例2中固体产品回收工序中目标物冻干后的hplc图谱;图3为实施例3中固体产品回收工序中目标物冻干后的hplc图谱;图4为实施例4中固体产品回收工序中目标物冻干后的hplc图谱;图5为比较例1中固体产品回收工序中目标物冻干前后的hplc图谱;图6为矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的质谱图;图7为矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的
1
h-nmr谱图;图8为矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的
13
c-nmr谱图。
[0024] 具体实施方式
[0025]
以下记载本发明的具体实施方式。
[0026]
本发明人经过广泛而深入地研究,首次研发出一种高纯度矢车菊-3-香豆酰-二葡
萄糖-5-葡萄糖苷对照品的制备方法。本发明的分离方法包括以下步骤:依次包括以下步骤:高压反相色谱柱分离工序,将含有矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的粗品上样至高压反相色谱柱,以5~15.0mpa压力,以含有0.05~1%的甲酸和15%-20%的乙腈的水进行洗脱,获得目标物的溶液。
[0027]
固体化工序,将反相色谱柱制备工序得到目标物的溶液,浓缩至矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的浓度为5%~50%,将浓缩液进行冷冻干燥,得到矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的甲酸盐的固体粉末。
[0028]
本发明的分离方法特别之处在于高压反相色谱柱分离工序和固体化工序的有机组合和协同,具体进行以下说明。
[0029]
所谓的高压反相色谱柱分离,通常指洗脱压力大于1mpa的色谱柱分离技术。本发明中,将目标物粗品浓缩液上样至高压反相色谱柱,以5-15.0mpa压力,以含有0.05~1%的甲酸和15%-20%的乙腈的水进行洗脱,获得目标物的溶液的工序。目标物可以根据高压色谱柱分离的紫外检测器的数据进行合并。一般目标物粗品在进行高压反相色谱柱分离工序之前已经具有90%的纯度,此步骤之后目标物的纯度可以达到98%以上。
[0030]
高压反相色谱柱分离工序中使用的洗脱剂为本发明的关键技术特征,其使用的洗脱剂非常重要。发明人曾尝试了不同的酸作为15%-20%的乙腈的添加剂,发现虽然在分离时各酸成分对分离结果影响较小,但是对最终获得的固体目标物的纯度影响非常大。
[0031]
具体而言,本发明人发现采用0.05~1%的甲酸的乙腈水溶液进行高压色谱柱分离后,在固体化工序中能够获得纯度98%以上的固体纯品。如果其他的酸,例如在花色苷对照品中最常见的盐酸盐所对应的盐酸。则无法获得高纯度的固体纯品。如果在上述高压色谱柱分离是采用盐酸作为洗脱剂的酸化剂,即使将盐酸浓度设定的非常低(后述实施例中验证了0.0075%(体积分数)的盐酸浓度),最后也难以通过上述固体化工序获得高纯度的固体纯品。这可以是矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷盐酸盐本身的稳定性较差造成的。
[0032]
总之,虽然目前花色苷类标准品多以盐酸盐的形式商品化提供,但是本发明人对矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的稳定性进行验证,发现其盐酸盐并不能稳定存在。盐酸盐冻干纯度明显降低,冻干前99%,冻干后最高纯度97%。矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的固体对照品,优选以甲酸盐的形式提供。
[0033]
高压色谱柱分离工序中,高压反相色谱柱的压力优选为8.0-10.0 mpa,此压力因为能平衡分离效果和分离速度而特别优选。对高压反相色谱柱的洗脱时使用的洗脱液,使用0.1~0.2%的甲酸和15%-20%的乙腈的水进行洗脱,有利于分离效果和速度,也有利于色谱柱的寿命,因此优选。
[0034]
在固体化工序中,将色谱柱制备工序得到目标物的溶液进行浓缩时,优选在45℃以下的温度进行减压除去乙腈后,浓缩过程控制在60分钟以内,然后直接进行冷冻干燥,这样可以提高稳定性,抑制纯品目标物的降解,获得更高纯度的固体对照品。不宜浓缩至浓度过高,因为担心洗脱液中的酸浓度提高会加剧目标物降解,优选的浓度应该是5%~30%,但是并不限于此。
[0035]
为了能更好的发挥上述分离方法的效果,整体上提供更高效的分离方案,优选的是通过依次包含以下工序的方法制备上述在高压反相色谱柱分离工序中作为原料的含有
矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的粗品:富集浓缩工序,以甘蓝红提取物为原料,利用酸化的醇类溶剂进行提取,浓缩得到第一原料浓缩液,将第一原料浓缩液上样至苯乙烯类骨架大孔树脂,用水洗脱去除杂质,再利用酸化的醇类溶剂洗脱获得含目标物的溶液,浓缩得到第二原料浓缩液的工序。
[0036]
色谱柱提纯工序,将第二原料浓缩液上样至中压反相色谱柱,以压力为0.05~0.5mpa进行梯度洗脱,洗脱过程为,以酸性的4%~5%乙腈的水溶液洗脱去除杂质,再以酸性的7%~9%乙腈的水洗脱收集该洗脱液浓缩得到第一粗品浓缩液;将第一粗品浓缩液上样至羟丙基葡聚糖凝胶色谱柱,利用酸性的含18~22%乙腈的水进行洗脱,利用hplc紫外检测器检测,根据吸收峰形收集样品溶液,浓缩干燥得到含有矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的粗品。
[0037]
上述富集浓缩工序,色谱柱提纯工序有机结合,能够快速提供适于本发明的目标物粗品。
[0038]
富集浓缩工序,可以甘蓝红提取物为原料,利用酸化的醇类溶剂进行提取,浓缩得到第一原料浓缩液,将第一原料浓缩液上样至苯乙烯类骨架大孔树脂,用水洗脱去除杂质,再利用酸化的醇类溶剂洗脱获得含目标物的溶液,浓缩得到第二原料浓缩液的工序,酸化的醇类溶剂例如可使用以体积比计含0.03~0.06%盐酸的甲醇或者乙醇。
[0039]
色谱柱提纯工序,将第二原料浓缩液上样至中压反相色谱柱,以压力为0.05~0.5mpa进行梯度洗脱,洗脱过程为,以酸性的4%~5%乙腈的水溶液洗脱去除杂质,再以酸性的7%~9%乙腈的水洗脱收集该洗脱液浓缩得到第一粗品浓缩液;将第一粗品浓缩液上样至羟丙基葡聚糖凝胶色谱柱,利用酸性的含18~22%乙腈的水进行洗脱,利用hplc紫外检测器检测,根据吸收峰形收集样品溶液,浓缩干燥得到粗品。
[0040]
本发明中,上述工序的组合和顺序也是非常重要的。
[0041]
富集和浓缩工序通过在常规的花色苷富集中通过不断的筛选和试验,发现利用苯乙烯-二乙烯苯骨架大孔树脂能够提高提取的收率,也有利于提高相应的纯度,具体原因尚不清楚,可能是由于苯乙烯-二乙烯苯骨架的大孔树脂的特定结构对于矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的吸附和洗脱特异性更强,发明人也尝试过苯乙烯-丙烯酸酯类的大孔树脂,发现收率上会比苯乙烯-二乙烯苯骨架大孔树脂差。因此在富集和浓缩步骤中,优选使用苯乙烯-二乙烯苯骨架。作为常用的苯乙烯-二乙烯苯骨架大孔树脂,可以使用ab-8、hpd-100、d-101等型号的大孔树脂等。
[0042]
在色谱柱提纯工序中,分为中压柱分离、凝胶分离两个步骤,他们的组合和先后顺序尤为重要。该步骤中,所谓中压柱分离,一般是指洗脱压力小于1mpa的柱色谱分离。本发明中,中压柱分离用于将与目标物极性有差异的其他成分分离,采用0.05~0.5mpa的洗脱压力能够实现分辨率和速度之间的平衡。对于中压柱的洗脱剂,以酸性的4%~5%乙腈的水溶液洗脱去除杂质,再以酸性的7%~9%乙腈的水洗脱收集该洗脱液浓缩得到第一粗品浓缩液,优选的是5%乙腈的水溶液洗脱去除杂质,再以酸性的8%乙腈的水洗脱收集该洗脱液浓缩得到第一粗品浓缩液。中压柱的中反相填料,可以用公知的非极性的,键合的官能团为烷烃的(例如:c18(ods)、c8、c4等)的硅胶。优选c18(ods)硅胶柱,即十八烷基硅烷键合硅胶,进一步优选粒径为40-60μm的十八烷基硅烷键合硅胶,能进一步平衡辨率和分离速度。酸性的乙腈的水溶液中,一般添加弱酸以提高分辨率,优选三氟乙酸,也可以使用甲酸。发明人
发现酸性的4%~5%乙腈的水溶液为含有0.03%~0.07%的三氟乙酸的乙腈的水溶液,酸性的7%~9%乙腈的水溶液为含有0.03%~0.07%的三氟乙酸的乙腈的水溶液时,分离的分辨率最好。此工序后能获得的目标物的纯度可以达到35~40%左右。中压柱分离工序中,中压反相色谱柱的压力优选为0.1mpa~0.2mpa,此压力因为能平衡分离效果和分离速度而特别优选。
[0043]
凝胶分离步骤即将第一粗品浓缩液上样至羟丙基葡聚糖凝胶色谱柱,利用酸性的含18~22%乙腈的水进行洗脱,利用hplc紫外检测器检测,根据吸收峰形收集样品溶液,浓缩得到第二粗品浓缩液的步骤。此工序为常规工序,但是发明人发现,其与中压柱分离的先后顺序非常重要,上述得到的第二原料浓缩液中,各成分紫外吸收状态类似,如果将凝胶工序至于中压分离之前,难以利用高效的紫外检测器根据吸收峰形收集样品溶液,几乎无法发挥凝胶分色谱柱分离的效果。此步骤中,酸性18~22%乙腈的水溶液为含有0.03%~0.07%的三氟乙酸的乙腈的水溶液时,分离的分辨率最好,因此优选。此工序后能获得的目标物的纯度可以达到80~90%左右。
[0044]
综上,本发明通过以上的制备方法,首次获得了固体纯度98%以上的的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷化合物。
[0045]
实施例
[0046]
以下,通过实施例进一步详细说明本发明的典型的提取方式。
[0047]
实施例1a.原料提取:将甘蓝红0.5kg粉末,加入5l含0.05%盐酸甲醇溶液,超声提取3次,每次1小时,冷却至室温,抽滤,合并滤液,45度浓缩至无醇味,共500ml;b.大孔树脂富集:称取8kg ab-8大孔吸附树脂装柱,将步骤a的浓缩液用大孔树脂动态吸附上样,然后用20l的水洗脱杂质,再用30l的0.05%盐酸甲醇溶液洗脱目标物质,收集洗脱液,浓缩至1l;c.中压制备:一根中压柱上样约固体含量100g粗品的浓缩液,目标含量约10g,平行上4根。
[0048]
中压制备柱:daiso rps c18,460
×
100 mm ,1.6-1.7kg填料。流速:70ml/min。花色苷样品溶解度较好,压力基本在0.1-0.2mpa。
[0049]
5%乙腈(含0.05%三氟乙酸)洗脱约4h,体积约20l除去前杂,8%乙腈(含0.05%三氟乙酸)洗脱出目标3-4h,体积约15l,浓缩得到36%左右矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷混合物粉末约60g,目标含量约25g;d.凝胶纯化:采用sephadex lh-20色谱柱,1100
×
55 mm,500g-600g填料。流速控制在2-3秒1滴;将步骤c所得到的目标粗品60g,用20%乙腈(含0.05%三氟乙酸)溶解上凝胶柱纯化,用20%乙腈(含0.05%三氟乙酸)洗脱,约500-600 ml,看到明显色带开始hplc跟踪检测,收集目标流份,经浓缩,得到纯度89%左右矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷(约150ml,浓缩后6g粉末,约4g目标);e.高压制备:将步骤d所得到的hplc 89%矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷,加
10ml 15%乙腈水溶解,一次上样约1.2g粉末(约含800mg目标化合物)。色谱柱采用ymc-triart c18,100
×
250 mm,5μm。流速控制为70ml/min,检测波长280/540nm,柱压控制在8.0-10.0mpa之间,利用15%-20%乙腈(含0.05%甲酸),线性梯度洗脱0-20min。约16-17min收集目标流份。得到hplc纯度98.4%矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷溶液;f.固体产品回收:将步骤e得到的高压流份45度浓缩20min、冷冻干燥,得纯度98.4%的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷固体0.50g。通过质谱、核磁共振图谱可以确认其结构。质谱与核磁的图谱请参照图6~图8。
[0050]
固体产品回收工序前目标物溶液的hplc谱图和冻干后固体的hplc图谱可以参照图1,能够稳定的提供目标品的高纯度甲酸盐。
[0051]
实施例2除了在e.高压制备步骤中,利用15%-20%乙腈(含0.1%甲酸)的洗脱液进行梯度洗脱,其余与实施例1同样的操作,得到纯度98.4%的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷约0.55g。固体产品回收工序前液体的hplc谱图,和冻干后固体的hplc图谱可以参照图2,能够稳定的提供目标品的高纯度甲酸盐。
[0052]
实施例3除了在e.高压制备步骤中,利用15%-20%乙腈(含0.5%甲酸)的洗脱液进行梯度洗脱,其余与实施例1同样的操作,得到纯度98.4%的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷约0.55g。固体产品回收工序前液体的hplc谱图,和冻干后固体的hplc图谱可以参照图3,能够稳定的提供目标品的高纯度甲酸盐。
[0053]
实施例4除了在e.高压制备步骤中,利用15%-20%乙腈(含1%甲酸)的洗脱液进行梯度洗脱,其余与实施例1同样的操作,得到纯度98.4%的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷约0.55g。固体产品回收工序前液体的hplc谱图,和冻干后固体的hplc图谱可以参照图4,能够稳定的提供目标品的高纯度甲酸盐。
[0054]
比较例1除了在e.高压制备步骤中,利用15%-20%乙腈(含0.075%盐酸)的洗脱液进行梯度洗脱,其余与实施例1同样的操作,得到纯度96.5%的矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷约0.45g。固体产品回收工序前液体的hplc谱图,和冻干后固体的hplc图谱可以参照图5。无法稳定的提供高纯度的目标品的盐酸盐,即使冷冻干燥也难以避免其降解。
[0055]
实施例4所使用的含1%甲酸的乙腈水溶液的酸度远远高于比较例使用的含0.075%盐酸的乙腈水溶液,提示矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的稳定性并非仅仅与酸度有关,应该是不同的阴离子部分对矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷稳定性存在较大影响。
[0056]
目前尚无矢车菊-3-香豆酰-二葡萄糖-5-葡萄糖苷的核磁图谱公开,本发明通过上述制备方法,获得了清晰度非常高的目标物的核磁图谱(参照图7和图8)。
[0057]
上述披露的各技术特征并不限于已披露的与其它特征的组合,本领域技术人员还可根据发明之目的进行各技术特征之间的其它组合,以实现本发明之目的,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种改进,均应落入本发明权利要求书确定的保护范围内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips