
 商标分类
商标分类  商标转让
商标转让 
一种吡非尼酮的合成方法与流程
 2021-02-02 09:02:58|
2021-02-02 09:02:58| 385|
385| 起点商标网
起点商标网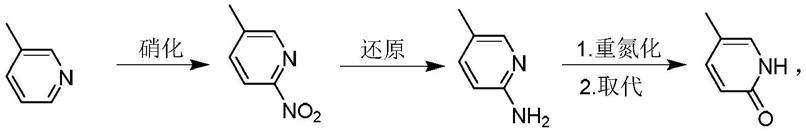
[0001]
本发明属于药物合成技术领域,具体涉及一种吡非尼酮的合成方法。
背景技术:
[0002]
吡非尼酮(pirfenidone)是一种新型广谱抗纤维化作用的吡啶酮类化合物,化学名称为5-甲基-1-苯基2-(1h)-吡啶酮,是一种白色或淡黄色结晶性粉末,能够防止纤维化和瘢痕的形成,也有一定的逆转化作用。该药是由日本盐野义于2008年上市,已获得美国食品药品监督管理局批准,是第一个通过重复、随机、安慰剂对照的-期临床试验证明对特发性肺纤维化(ipf)有一定疗效的药物;且该药在肾间质纤维化、肝纤维化等纤维化疾病也有较好疗效;同时还在肾脏疾病(局灶性节段性肾小球硬化症)、肥厚性心肌病、成年人i型多发性神经纤维瘤、青少年i型多发性神经纤维瘤和丛状神经纤维瘤、糖尿病伴有肾脏疾病、子宫平滑肌瘤的-期临床研究等方面有广泛应用。
[0003]
吡非尼酮目前已知的合成方法大多采用5-甲基-2(h)-吡啶酮作为起始原料,和碘苯在催化剂和缚酸剂的作用下进行高温反应,现有技术报道的收率(wo2008147170, wo2008157786,wo2010141600,wo2012122165,wo2015112701,cn109593060, cn111039857,cn1817862,cn102241625,cn102558040)大都为55-85%之间,反应式如下式:
[0004]
其中主要原料5-甲基-2(h)-吡啶酮的价格较为昂贵,而且不易获得,需从3-甲基吡啶为起始原料,通过3-4步合成反应才能获得,如下式:收率中等;而其中另外一个主要反应试剂碘苯的价格也较为昂贵,反应得到的产品纯度仍不理想,反应后处理与成品精制过程的损失较大。
[0005]
已公开专利方法(wo2003014087,wo2010141600,wo2016122420, wo2017130166,wo201741011006,wo2018083709,wo2018178996,wo20180334434, ep3266767,us20180009753,cn101891676,cn107663168)对碘苯试剂进行改进,采用溴苯来替代碘苯进行反应,成本有一定的降低,但收率也有一定程度的下降,仍不理想。同样,也有专利方法(wo2017072216)采用价格更为廉价的氯苯作为替代,其结果是造成反应温度大幅度升高,反应时间延长,收率也下降较多。
[0006]
另有专利(us20140094456,wo2015153683)采用同样价格昂贵的苯硼酸替代碘苯进行反应,收率仍不理想,且成本较高。
[0007]
因此,亟需研发一种原料廉价易得、工艺方法简单可行的吡非尼酮合成方法。
技术实现要素:
[0008]
针对上述现有技术关于吡非尼酮合成方法存在原料成本较高,原料合成路线复杂,来源不易得;合成过程中不可避免的采用较为昂贵的试剂,如碘苯,苯硼酸等问题,本发明的目的是提供一种原料廉价易得,工艺方法简单可行的吡非尼酮的合成方法,所述吡非尼酮的合成路线如图1所示。
[0009]
为实现上述目的,本发明采用以下技术方案。
[0010]
一种吡非尼酮的合成方法,包括步骤如下。
[0011]
步骤1、于反应器1中加入溶剂a,置换氮气,加入原料苯胺,开启搅拌,缓慢加入 2-甲基-1,3-丙二醛,控制温度和加料时间,加完后,再缓慢加入缩合剂a,控制加料时间,监测原料苯胺反应情况,反应完毕停止反应,得到中间体1的溶液。
[0012]
步骤2、将步骤1制得的中间体1的溶液冷却,加入缚酸剂a,控制温度,再加入催化剂a,加完继续搅拌,再缓慢滴入酰化剂a,控制温度和加料时间,滴加完毕;再升温继续反应,反应完毕后降温,过滤,滤饼用溶剂a淋洗,合并滤液,得到中间体2的溶液。
[0013]
步骤3、将步骤2制得的中间体2的溶液减压蒸干,加入溶剂b并转移至反应器2,置换氮气,开启搅拌,加入缩合剂b,控制反应温度和反应时间,反应完毕,将反应液减压浓缩,降温析晶,过滤,用溶剂c淋洗,得到吡非尼酮粗品。
[0014]
步骤4、将步骤3制得的吡非尼酮粗品,加入溶剂c加热,再冷却析晶,过滤,用溶剂d淋洗,减压干燥,得到吡非尼酮成品。
[0015]
进一步地,所述吡非尼酮的合成方法,具体包括步骤如下。
[0016]
步骤1、以苯胺作为起始原料,于反应器1中加入溶剂a,置换氮气,加入原料苯胺,开启搅拌,控制转速120rpm至400rpm,缓慢加入2-甲基-1,3-丙二醛,维持温度范围 40℃至85℃,控制在1h至4h加完,再缓慢加入缩合剂a,控制在1h至4h加完,继续反应2h至16h,监测原料苯胺反应情况,反应完毕停止反应,,得到中间体1的溶液。
[0017]
步骤2、将步骤1制得的反应液冷却至-40℃至15℃,加入缚酸剂a,控制温度不超过-20℃至15℃,再加入催化剂a,加完继续搅拌10min至30min,再缓慢滴入酰化剂a,控制温度不超过-10至60℃,控制时间2h至8h滴加完毕。再升温至0℃至回流温度继续反应2h至4h,监测中间体1是否反应完成,若未反应完则补加一定量的酰化剂a继续至反应完成。降温至0℃至室温,过滤,滤饼用溶剂a淋洗,合并滤液,得到中间体2的溶液。
[0018]
步骤3、将步骤2制得的中间体2的溶液减压蒸干,得到淡黄色至红棕色液体,加入溶剂b并转移至反应器2,置换氮气,开启搅拌,控制反应温度在15℃至80℃,加入缩合剂b,再将温度控制在15℃至80℃反应2h至8h,监测中间体2剩余量<0.5%,停止反应,将反应液减压浓缩至溶质质量的2倍至5倍,将温度降至-20℃至25℃,保持温度析晶 2h至6h,过滤,用溶剂c淋洗,得到吡非尼酮粗品,总收率约83-86%。
[0019]
步骤4、将步骤3制得的吡非尼酮粗品,用固体质量2倍至10倍溶剂c加热到40℃至120℃,维持0.5h至2h,再冷却至25℃至-25℃,维持1h至4h析晶,过滤,用0.5倍至 4倍体积的溶剂d淋洗,减压干燥,温度25℃至110℃,压力101kpa至0.1kpa,时间2h 至8h,得到吡非尼酮成品,纯度99.9%以上,无>0.05%单杂。
[0020]
进一步地,所述的苯胺与2-甲基-1,3-丙二醛、溶剂a、和缩合剂a、缚酸剂a、催化剂a、酰化剂a、溶剂b与缩合剂b的摩尔比为1:(1~3):(3~8):(1~3):(1~3): (0.01~0.2):
(1~2):(3~10):(1~3)。
[0021]
进一步地,步骤1中所述的溶剂a为n,n-二甲基乙酰胺、甲苯、二氯甲烷、乙酸乙酯、四氢呋喃中的一种或几种的混合物。
[0022]
进一步地,步骤1中所述的缩合剂a为1-(1-哌啶基)环己烯、n,n-二甲基甲酰胺二甲基缩醛、原甲酸三乙酯中的一种或几种的混合物。
[0023]
优选的,步骤1中2-甲基-1,3-丙二醛和苯胺的摩尔比为(1~2):1。
[0024]
优选的,步骤1中所述的反应温度为30℃至65℃。
[0025]
进一步地,步骤2中所述的溶剂a为n,n-二甲基乙酰胺、甲苯、二氯甲烷、乙酸中的一种或几种的混合物。
[0026]
进一步地,步骤2中所述的缚酸剂a为碳酸钾、氧化钙、或三乙胺吡啶。
[0027]
进一步地,步骤2中所述的酰化剂a为乙酰氯或乙酸酐。
[0028]
进一步地,步骤2中所述的催化剂a为4-二甲氨基吡啶、三甲基氯硅烷或浓硫酸。
[0029]
进一步地,步骤3中所述的溶剂b为四氢呋喃、乙醇、甲苯、n,n-二甲基甲酰胺中的一种或几种的混合物。
[0030]
进一步地,步骤3中缩合剂b为1-(1-哌啶基)环己烯、n,n-二甲基甲酰胺二甲基缩醛、原乙酸三甲酯中的一种或几种的混合物。
[0031]
优选的,步骤3中所述的反应温度优选为45℃至65℃。
[0032]
优选的,步骤3中所述的析晶温度优选为-10℃至5℃。
[0033]
进一步地,步骤4中所述溶剂c为n,n-二甲基乙酰胺、甲苯、乙醇、异丙醇、乙酸乙酯中的一种或几种的混合物。
[0034]
优选的,步骤4中所述溶剂c的加入质量为固体质量的3倍至6倍。
[0035]
优选的,步骤4中所述加热温度为60℃至100℃。
[0036]
优选的,步骤4中所述析晶时间为2h至4h。
[0037]
进一步地,步骤4中所述溶剂d为乙醇、异丙醇、乙酸、正庚烷中的一种或几种混合物。
[0038]
优选的,步骤4中所述干燥温度为60℃至80℃。
[0039]
优选的,步骤4中所述干燥压力优选为10kpa至0.1kpa。
[0040]
其中原料2-甲基-1,3-丙二醛的来源,可通过丙醛作为原料,与甲酸乙酯在碱的催化下缩合得到,如图2所示。
[0041]
本发明中提到的专业词汇与相关领域技术人员的普遍认知相一致,如:当量表示与基准物物质的量的摩尔比例;干燥条件的压力为绝对压力值而非真空度;h表示小时,min 表示分钟;收率为摩尔收率。如有冲突,以本说明书解释为准。
[0042]
与现有技术比,本发明的有益效果如下。
[0043]
本发明提供的的吡非尼酮合成方法,相比于已公开的合成技术路线,原料廉价易得,方法简单可行,反应选择性好,合成路线总收率高,产品易纯化,适合药品工业化生产应用。
附图说明
[0044]
图1是吡非尼酮的合成路线。
[0045]
图2是原料2-甲基-1,3-丙二醛的合成路线。
具体实施方式
[0046]
下面结合附图和实施例对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但这些实施例仅为了对本发明加以说明,本发明并不限于这些内容。
[0047]
实施例1中间体1的制备。
[0048]
于反应器1中加入400mln,n-二甲基乙酰胺,置换氮气,加入原料100g苯胺,开启搅拌,控制转速240rpm,缓慢加入1.25当量的2-甲基-1,3-丙二醛,维持温度60℃至65℃,控制在1.5h加完,再缓慢加入1.3倍当量n,n-二甲基甲酰胺二甲基缩醛,控制在2h加完,继续反应4h,原料苯胺反应完毕,停止反应,得到中间体1的溶液。
[0049]
实施例2中间体1的制备。
[0050]
于反应器1中加入400ml甲苯,置换氮气,加入原料100g苯胺,开启搅拌,控制转速260rpm,缓慢加入1.2倍当量的2-甲基-1,3-丙二醛,维持温度80-85℃,控制在1h加完,再缓慢加入1.0倍当量原甲酸三乙酯,控制在1h加完,继续反应5h,原料苯胺反应完毕,停止反应,得到中间体1的溶液。
[0051]
实施例3中间体1的制备。
[0052]
于反应器1中加入300ml二氯甲烷,置换氮气,加入原料100g苯胺,开启搅拌,控制转速280rpm,缓慢加入2倍当量的2-甲基-1,3-丙二醛,维持温度38℃-41℃,控制在1h 加完,再缓慢加入1.2倍当量1-(1-哌啶基)环己烯,控制在1.5h加完,继续反应4h,原料苯胺反应完毕,停止反应,得到中间体1的溶液。
[0053]
实施例4中间体1的制备。
[0054]
于反应器1中加入400ml乙酸乙酯,置换氮气,加入原料100g苯胺,开启搅拌,控制转速240rpm,缓慢加入1.3倍当量的2-甲基-1,3-丙二醛,维持温度70℃至75℃,控制在 1.5h加完,再缓慢加入1.1倍当量原甲酸三乙酯,控制在1h加完,继续反应4h,原料苯胺反应完毕,停止反应,得到中间体1的溶液。
[0055]
实施例5中间体1的制备。
[0056]
于反应器1中加入400ml四氢呋喃,置换氮气,加入原料100g苯胺,开启搅拌,控制转速240rpm,缓慢加入1.2倍当量的2-甲基-1,3-丙二醛,维持温度60℃至65℃,控制在 1h加完,再缓慢加入1.5倍当量n,n-二甲基甲酰胺二甲基缩醛,控制在1.5h加完,继续反应6h,原料苯胺反应完毕,停止反应,得到中间体1的溶液。
[0057]
实施例6中间体2的制备。
[0058]
将中间体1的n,n-二甲基乙酰胺溶液冷却至0℃至5℃,加入1.5倍当量碳酸钾,控制温度不超过5℃,再加入0.05倍当量4-二甲氨基吡啶,加完继续搅拌10min,再缓慢滴入 1.1倍当量乙酰氯,控制温度不超过5℃,控制时间2h滴加完毕,再升温至40℃继续反应 2h,反应完成。降温至室温至0℃,过滤,滤饼用n,n-二甲基乙酰胺淋洗,合并滤液,得到中间体2的溶液。
[0059]
实施例7中间体2的制备。
[0060]
将中间体1的甲苯溶液冷却至-10℃至0℃,加入1.5倍三乙胺,控制温度不超过0℃,再加入0.1倍当量三甲基氯硅烷,加完继续搅拌20min,再缓慢滴入1.1倍当量乙酸酐,控
制温度不超过0℃,控制时间2.5h滴加完毕。再升温至60℃继续反应4h,反应完成。降温至室温至0℃,过滤,滤饼用甲苯淋洗,合并滤液,得到中间体2的溶液。
[0061]
实施例8中间体2的制备。
[0062]
将中间体1的二氯甲烷溶液冷却至-5℃至0℃,加入1.5倍当量吡啶,控制温度不超过5℃,再加入0.02倍当量4-二甲氨基吡啶,加完继续搅拌15min,再缓慢滴入1.1倍当量乙酰氯,控制温度不超过5℃,控制时间1.5h滴加完毕。再升温至40℃继续反应4h,反应完成。降温至室温至0℃,过滤,滤饼用二氯甲烷淋洗,合并滤液,得到中间体2的溶液。
[0063]
实施例9中间体2的制备。
[0064]
将中间体1的乙酸乙酯溶液冷却至0℃-5℃,加入1.2倍当量氧化钙,控制温度不超过5℃,再加入0.05倍当量4-二甲氨基吡啶,加完继续搅拌约10min,再缓慢滴入1.05倍当量乙酰氯,控制温度不超过5℃,控制时间2.5h滴加完毕。再升温至65℃继续反应2h,反应完成。降温至室温至0℃,过滤,滤饼乙酸乙酯淋洗,合并滤液,得到中间体2的溶液。
[0065]
实施例10中间体2的制备。
[0066]
将中间体1的四氢呋喃溶液冷却至-5℃至0℃,加入1.2倍当量三乙胺,控制温度不超过5℃,再加入0.01倍当量浓硫酸,加完继续搅拌10min,再缓慢滴入1.2倍当量乙酸酐,控制温度不超过0℃,控制时间2h滴加完毕。再升温至65℃继续反应4h,反应完成。降温至室温至0℃,过滤,滤饼用四氢呋喃淋洗,合并滤液,得到中间体2的溶液。
[0067]
实施例11吡非尼酮的制备。
[0068]
将中间体2的溶液减压蒸干,加入500ml四氢呋喃并转移至反应器2,置换氮气,开启搅拌,控制反应温度在35℃至40℃,加入1.5倍当量n,n-二甲基甲酰胺二甲基缩醛,再将温度控制在60℃至65℃反应4h,中间体2剩余量<0.3%,停止反应,将反应液减压浓缩至溶质质量的3.5倍,将温度降至-15℃至-10℃,保持温度析晶4h,过滤,用100ml乙醇分2次淋洗,得到吡非尼酮粗品,收率84.5%。
[0069]
实施例12吡非尼酮的制备。
[0070]
将中间体2的溶液减压蒸干,加入450ml甲苯并转移至反应器2,置换氮气,开启搅拌,控制反应温度在55℃至60℃,加入原乙酸三甲酯,再将温度控制在85℃至90℃反应 6h,中间体2剩余量<0.25%,停止反应,将反应液减压浓缩至溶质质量的4倍,将温度降至-10℃至-5℃,保持温度析晶3h,过滤,用100ml异丙醇分2次淋洗,得到吡非尼酮粗品,收率83.7%。
[0071]
实施例13吡非尼酮的制备。
[0072]
将中间体2的溶液减压蒸干,加入400ml无水乙醇并转移至反应器2,置换氮气,开启搅拌,控制反应温度在25℃至30℃,加入加入原乙酸三甲酯,再将温度控制在75℃至 80℃反应8h,中间体2剩余量<0.5%,停止反应,将反应液减压浓缩至溶质质量的4倍,将温度降至-10℃至-5℃,保持温度析晶4h,过滤,用100ml无水乙醇淋洗,得到吡非尼酮粗品,收率88.3%。
[0073]
实施例14吡非尼酮的精制。
[0074]
将吡非尼酮粗品,用固体质量4倍无水乙醇加热到75℃至80℃,维持1h,再冷却至
ꢀ-
5℃,维持析晶4h,过滤,用0.5体积的正庚烷淋洗,减压干燥,温度65℃,压力 0.3kpa,时间4h,得到吡非尼酮成品,精制收率92.5%,纯度99.92%。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips