
 商标分类
商标分类  商标转让
商标转让 
一种(S)-磷酸氯喹的不对称合成方法与流程
 2021-02-02 08:02:14|
2021-02-02 08:02:14| 326|
326| 起点商标网
起点商标网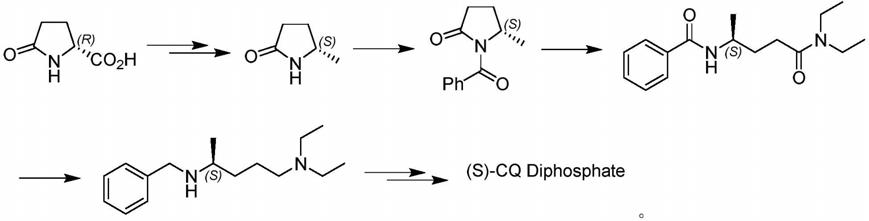
一种(s)-磷酸氯喹的不对称合成方法
技术领域
[0001]
本发明涉及一种(s)-磷酸氯喹的不对称合成方法,属于药物合成领域。
背景技术:
[0002]
发明人调查了磷酸氯喹的相关资料。氯喹是治疗疟疾的传统老药,之后也被应用于治疗类风湿关节炎、系统性红斑狼疮等自身免疫性疾病,但在临床使用中的毒副作用较重,尤其是对于心脏的毒性是致命的。百度百科中关于磷酸氯喹的词条内容显示,氯喹分子中喹啉环的4位氨基所连接的饱和碳原子应该是一个具有手性中心的碳原子,而临床上使用的磷酸氯喹却是一个外消旋体。在发明人正在参加学习的高中化学奥林匹克竞赛的培训课程中,授课老师和相关竞赛的参考书比较详细讲解了关于手性分子和手性药物的知识。对于手性分子的对映异构体而言,它们在非手性环境中的物理性质和化学性质基本上是相同的,但在手性环境中,它们的性质是不相同的。生物体内的酶和蛋白质等都是具有手性的,所以对映异构体的生理活性往往有很大的差异。比如,左旋尼古丁的毒性比右旋尼古丁的毒性大很多;左旋氯霉素有疗效,而右旋氯霉素却没有疗效;左旋香芹酮的香气和右旋异构体的香气并不相同。历史上,因为手性药物造成的悲剧,最典型的就是“反应停”事件。1953年,联邦德国上市了一种可以治疗孕妇呕吐的药物——沙利度胺。两年后,欧洲地区陆续发现了1万2千多名畸形婴儿。研究结果发现,沙利度胺的r构型具有镇静止吐作用,而s构型异构体却是致畸的罪魁祸首。1984年,荷兰药理学家阿里安斯极力提倡手性药物以单一对映体上市,抨击以消旋体形式进行药理研究和上市。1992年,美国药品监管部门规定,新的手性药物上市之前必须分别对手性对映异构体进行药效和毒性研究,否则不允许上市。2006年,我国药监局也出台了相应的政策法规。
[0003]
通过检索文献资料,我们得到已有一些研究推荐对氯喹和羟氯喹的手性异构体进行毒性对比研究,以确定毒性相对较低的手性异构体来对新冠病毒进行临床治疗(moleculars,2020, 25,1834;drug discovery today,2020,25,1121-1123;biorxiv,doi: https://doi.org/10.1101/2020.05.26.114033.)。根据已有的研究资料,这些文献还特别推荐s构型的氯喹可能是相对更优的手性异构体,可能具有较低的心脏和视网膜毒性(drug discoverytoday,2020,25,1121-1123;biorxiv,doi:https://doi.org/10.1101/2020.05.26.114033.)。于是,我们着手开展了合成(s)-磷酸氯喹的研究。
[0004]
(s)-磷酸氯喹或(s)-氯喹的制备,最早报道的方法是拆分法(journal of the americanchemical society,1949,71,1129-1130;chemischeberichte,1978,111,2732-2734;chirality,1993, 5,188-190)。
[0005]
gideon blauer等人报道了采用(s)或(r)-焦谷氨酸为手性源物料,经过10步反应来制备(s) 或(r)-磷酸氯喹(chirality,1998,10,556-563)。合成路线如下:
[0006][0007]
manish sinha等人报道了以(s)-丙氨酸为手性源,经过12步反制备了(s)-氯喹 (bioorganic&medicinal chemistry,2014,22,5950-5960)。合成路线如下:
[0008][0009]
j.cymerman craig等以l-谷氨酸二乙酯为手性源,经过10步反应,合成得到(r)-5-二乙基氨基-2-戊胺,继而与4,7-二氯喹啉缩合,得到(r)-氯喹(journal of organic chemistry,1988, 53,1167-1170)。如果以d-谷氨酸二乙酯为手性源,采用相同的路线,即可得到(s)-氯喹。
[0010]
以上采用不同的手性源为起始物料来合成制备(s)-氯喹/磷酸氯喹的方法,显然合成路线很长,成本很高,不适合大规模工业化生产。
[0011]
陈福欣等以4-氨基-7-氯喹啉和5-二乙基氨基-2-戊酮为原料,在手性酸催化剂的催化下,通过不对称还原胺化反应,得到(s)-氯喹。合成路线如下:
[0012][0013]
以上不对称催化的合成方法,虽然合成路线很简单,但由于4-氨基-7-喹啉上的氨基为芳香胺,亲核进攻能力较差,因此该方法提供的(s)-氯喹的收率较低(36.5%~68%),不对称催化的手性选择性也不是非常理想,产物(s)-氯喹的ee值为68%~92%。
技术实现要素:
[0014]
本发明的目的在于提供一种(s)-磷酸氯喹的不对称合成新方法,旨在克服以上制
备方法中存在的不足。
[0015]
磷酸氯喹的工业化生产工艺是以4,7-二氯喹啉为母核、5-(n-二乙基氨基)-2-戊胺为侧链,在高温下,经过亲核取代反应,得到氯喹,再与磷酸成盐,得到磷酸氯喹。母核4,7-二氯喹啉的合成是以3-氯苯胺和丁酮二酸二乙酯为原料,经过缩合、环合、水解、脱羧后再氯代来制备的。侧链5-(n-二乙基氨基)-2-戊胺的合成是以乙酰乙酸乙酯和2-二乙基氨基氯乙烷为原料,经过亲核取代、水解脱羧后再经还原胺化反应来制备的。合成路线如下:
[0016][0017]
结合以上氯喹的合成路线和方法,发明人考虑,如果可以很方便地合成制备s构型的侧链,即(s)-5-(n-二乙基氨基)-2-戊胺,则可以采用原合成路线方法,很方便的制备得到s构型的氯喹。
[0018]
通过检索文献,发明人发现手性辅助试剂(r)/(s)-α-甲基苄胺在手性胺的不对称合成中应用得十分广泛,该方法稳定可靠,手性选择性高(上海大学学报(自然科学版),2008,14,80-84; synthesis,2013,45,153-166)。(r)/(s)-α-甲基苄胺是市场上非常易得的医药化工中间体,且价格低廉,很适合工业化制备手性胺。在该方法中,常采用路易斯酸催化,且(r)/(s)-α-甲基苄胺可以分别确定地得到(r)/(s)-胺(journal of organic chemistry,2008,73,1297-1305;organicletters,2005,7,4967-4970)。
[0019]
由于市场上有5-二乙基氨基-2-戊酮和4,7-二氯喹啉化工中间体供应,因此,本发明采用 5-二甲氨基-2-戊酮、(s)-α-甲基苄胺和4,7-二氯喹啉为原料,在路易斯酸的存在下,通过不对称还原胺化反应,得到(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺,再利用催化加氢脱苄,得到(s)-5-二乙基氨基-2-戊胺,继而与4.7-二氯喹啉缩合、与磷酸成盐,得到(s)-磷酸氯喹。合成路线如下:
[0020][0021]
本发明提供一种(s)-磷酸氯喹的不对称合成制备方法,其制备方法步骤如下:
[0022]
步骤1:以5-二甲氨基-2-戊酮和(s)-α-甲基苄胺为原料,在路易斯酸的存在下,通过不对称还原胺化反应,得到(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺;
[0023]
步骤2:(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺在催化加氢条件下,脱去苄基,得到(s)-5-二乙基氨基-2-戊胺;
[0024]
步骤3:(s)-5-二乙基氨基-2-戊胺和4,7-二氯喹啉缩合,得到(s)-氯喹;
[0025]
步骤4:(s)-氯喹与磷酸成盐、重结晶,得到(s)-磷酸氯喹。
[0026]
步骤1中所述的路易斯酸可选择钛酸异丙酯、硼酸三异丙酯、异丙醇铝、乙酸镱、乙酸铈中的一种或多种混合试剂。
[0027]
步骤1中所述的路易斯酸和5-二乙基氨基-2-戊酮的投料摩尔比为0.5~1.5:1。
[0028]
步骤1中所述的还原胺化反应所采用的还原试剂可以是催化加氢、硼氢化钠、氰基硼氢化钠、三乙酰氧基硼氢化钠中的一种。
[0029]
本发明所提供的一种(s)-磷酸氯喹的不对称合成制备方法,其创新性体现在以下几点:
[0030]
(1)采用市场上易得的5-二甲氨基-2-戊酮、(s)-α-甲基苄胺和4,7-二氯喹啉为原料,仅经过4步反应,即可得到目标产品,反应步骤少,手性选择性和收率高;
[0031]
(2)合成工艺无特殊苛刻的反应条件,工艺稳定性好,可靠性高,经济高效,非常适合大规模工业化生产。
具体实施方式
[0032]
以下典型实施例用来举例说明本发明,在本领域内的技术人员对本发明所做的简单替换或改进等均属于本发明所保护的技术方案之内。
[0033]
实施例1:(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺的合成
[0034]
反应式如下:
[0035][0036]
在500ml氢化反应釜中加入甲醇(150ml),开启搅拌,再依次加入5-二乙基氨基-2-戊酮(15.7g,0.1mol)、(s)-甲基苄胺(12.7g,0.105mol)和钛酸异丙酯(14.2g,0.05mol)。在室温下搅拌半小时后,加入湿的raney-ni(25g),用氢气置换三次后,通入氢气至1.0mpa,继续在室温下搅拌反应12小时。反应结束后,放出釜内氢气,打开反应釜,加入naoh溶液
(1.0m, 100ml),继续在室温下搅拌1小时,反应体系成混悬液状。过滤,滤液用旋转蒸发仪蒸除约一半的溶剂后,加入二氯甲烷萃取三次(60ml
×
3)。合并二氯甲烷有机相,用无水硫酸钠干燥后,过滤,蒸除二氯甲烷溶剂,得到无色油状液体22.1g(0.0842mol),收率84.2%。
[0037]
实施例2:(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺的合成
[0038]
反应式如下:
[0039][0040]
在500ml氢化反应釜中加入甲醇(150ml),开启搅拌,再依次加入5-二乙基氨基-2-戊酮(15.7g,0.1mol)、(s)-甲基苄胺(12.7g,0.105mol)和钛酸异丙酯(28.4g,0.1mol)。在室温下搅拌半小时后,加入湿的raney-ni(25g),用氢气置换三次后,通入氢气至1.0mpa,继续在室温下搅拌反应12小时。反应结束后,放出釜内氢气,打开反应釜,加入naoh溶液(1.0m, 100ml),继续在室温下搅拌1小时,反应体系成混悬液状。过滤,滤液用旋转蒸发仪蒸除约一半的溶剂后,加入二氯甲烷萃取三次(60ml
×
3)。合并二氯甲烷有机相,用无水硫酸钠干燥后,过滤,蒸除二氯甲烷溶剂,得到无色油状液体25.2g(0.096mol),收率96%。
[0041]
实施例3:(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺的合成
[0042]
反应式如下:
[0043][0044]
在500ml氢化反应釜中加入甲醇(150ml),开启搅拌,再依次加入5-二乙基氨基-2-戊酮(15.7g,0.1mol)、(s)-甲基苄胺(12.7g,0.105mol)和钛酸异丙酯(42.6g,0.15mol)。在室温下搅拌半小时后,加入湿的raney-ni(25g),用氢气置换三次后,通入氢气至1.0mpa,继续在室温下搅拌反应12小时。反应结束后,放出釜内氢气,打开反应釜,加入naoh溶液(1.0m, 100ml),继续在室温下搅拌1小时,反应体系成混悬液状。过滤,滤液用旋转蒸发仪蒸除约一半的溶剂后,加入二氯甲烷萃取三次(60ml
×
3)。合并二氯甲烷有机相,用无水硫酸钠干燥后,过滤,蒸除二氯甲烷溶剂,得到无色油状液体25.6g(0.0976mol),收率97.6%。
[0045]
实施例4:(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺的合成
[0046]
反应式如下:
[0047][0048]
在500ml反应瓶中加入甲醇(150ml),开启搅拌,再依次加入5-二乙基氨基-2-戊酮(15.7g, 0.1mol)、(s)-甲基苄胺(12.7g,0.105mol)和硼酸三异丙酯(22.5g,0.12mol)。在室温下搅拌半小时后,缓慢分批加入硼氢化钠(3.8g,0.1mol),继续在室温下搅拌反应12小时。反应结束后,加入饱和nh4cl溶液(100ml),继续在室温下搅拌1小时。过滤,滤液用旋转蒸发仪蒸除约一半的溶剂后,加入二氯甲烷萃取三次(60ml
×
3)。合并二氯甲烷有机相,用无水硫酸钠干燥后,过滤,蒸除二氯甲烷溶剂,得到无色油状液体23.9g(0.0911mol),收率
91.1%。
[0049]
实施例5:(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺的合成
[0050]
反应式如下:
[0051][0052]
在500ml反应瓶中加入甲醇(150ml),开启搅拌,再依次加入5-二乙基氨基-2-戊酮(15.7g, 0.1mol)、(s)-甲基苄胺(12.7g,0.105mol)和新鲜干燥的无水乙酸镱(28.0g,0.08mol)。在室温下搅拌半小时后,缓慢分批加入氰基硼氢化钠(12.5g,0.2mol),继续在室温下搅拌反应12 小时。反应结束后,加入饱和nh4cl溶液(100ml),继续在室温下搅拌1小时。过滤,滤液用旋转蒸发仪蒸除约一半的溶剂后,加入二氯甲烷萃取三次(60ml
×
3)。合并二氯甲烷有机相,用无水硫酸钠干燥后,过滤,蒸除二氯甲烷溶剂,得到无色油状液体24.5g(0.0933mol),收率93.3%。
[0053]
实施例6:(s)-5-(n-二乙基氨基)-2-戊胺的合成
[0054]
反应式如下:
[0055][0056]
在500ml氢化反应釜中依次加入5%的pd/c(10g)、实施例2所得的(s,s)-5-(n
’-
二乙基氨基)-n-((1-苯基)乙基)-2-戊胺(25.2g,0.096mol)和甲醇(100ml),开启搅拌,用氢气置换三次后,通入氢气至0.5mpa,在室温下搅拌反应24小时。反应结束后,放出釜内氢气,打开反应釜,反应液过滤后,蒸除甲醇溶剂,得到无色油状液体15.1g(0.095mol),收率99%。
[0057]
实施例7:(s)-氯喹的合成
[0058]
反应式如下:
[0059][0060]
在250ml反应瓶中加入苯酚(100g),加热到100℃使苯酚熔融,在开启搅拌下加入4,7
-ꢀ
二氯喹啉(18.8g,0.095mol),待固体溶解成均相后,加入实施例6所得的(s)-5-(n-二乙基氨基)-2-戊胺(15.1g,0.095mol)。再将反应液加热到135℃,反应3小时。反应结束后,减压蒸除大部分苯酚后降温至室温,所得固体用二氯甲烷(200ml)溶解,再加入1.0m的naoh溶液洗涤三次(100ml
×
3),再用纯水洗涤二次(100ml
×
2)。有机相用无水硫酸钠干燥,过滤后减压蒸除溶剂,冷却(0~4℃)析晶,得到黄色(s)-氯喹粗品。粗品用正己烷重结晶后得到淡黄色(s)-氯喹26.1g(0.0816mol),收率85.9%,
[0061]
所得(s)-氯喹的对映异构体含量分析采用高效液相色谱法,检测条件为:采用
ay-h手性柱;流动相为正己烷/异丙醇/二乙胺=85:15:0.1(v/v/v);流速为 0.8ml/min;柱温为35℃;检测波长为254nm。分析检测结果为:(r)-氯喹未检出。因此,(s)
-ꢀ
氯喹和(s)-磷酸氯喹的ee值应大于99%。
[0062]
实施例8:(s)-磷酸氯喹的合成
[0063]
反应式如下:
[0064][0065]
在250ml反应瓶中加入甲醇(100ml)和实施例7所得的(s)-氯喹(25g,0.0782mol),加热至回流,滴加85%的浓磷酸(10.5g,0.091mol)。滴加完毕后,继续在回流状态下搅拌1小时。在保持缓慢搅拌的状态下,缓慢降温至室温。12小时后,过滤,滤饼用纯水重结晶,得到白色结晶性固体36.3g(0.0704mol),收率90%,1h nmr(d2o, 500mhz):δ(ppm)1.01~1.06(m,6h),1.18(d,3h),1.54~1.60(m,4h),2.92~2.99(m,6h), 3.80~3.84(m,1h),6.51(d,1h),7.03(dd,1h),7.13(s,1h),7.65(d,1h),7.94(d,1h)。
13
c nmr (d2o,125mhz):δ(ppm)8.00,8.06,18.55,20.16,31.70,46.99,47.07,49.41,51.03,98.49,114.39, 118.32,123.67,126.77,137.34,138.69,141.98,154.59。hrms(esi
+
):c
18
h
27
cln3[m+h]
+
(m/z):理论值为320.1888,实测值为320.1886。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips