
 商标分类
商标分类  商标转让
商标转让 
具有增强的蛋白质表达效率的宿主细胞及其用途的制作方法
 2021-02-02 08:02:26|
2021-02-02 08:02:26| 412|
412| 起点商标网
起点商标网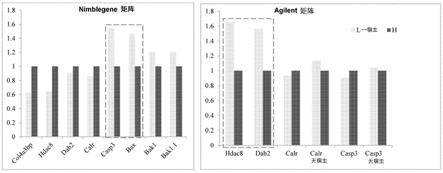
[0001]
本发明涉及用于蛋白质生产的宿主细胞,尤其涉及经工程改造的宿主细胞,如cho细胞,该细胞相较于野生型细胞可以产生较高水平的蛋白质。
背景技术:
[0002]
蛋白质药剂典型地是通过在适合的宿主细胞中表达所产生的。中国仓鼠卵巢(cho)细胞为用于蛋白质药物生产的最常使用的宿主细胞。为了提高蛋白质药物生产效率,正在开发研究宿主细胞的优化(例如通过修饰宿主细胞的基因)或下游制备方法的优化。目前多种策略可用于增强蛋白质表达和/或分泌,例如通过使用化学试剂或通过修饰细胞的基因。
[0003]
在基因修饰的情况下,典型地是靶向涉及转录、转录后调节、翻译及翻译后事件的基因。例如,转录后调节组件(ptre)业已成操作的标的以提高蛋白质表达。(参见mariati等人,“转录后调控元件,用于增强哺乳动物细胞中瞬时基因的表达水平”,分子生物学方法,2012,801:125-35。)
[0004]
尽管用于调节蛋白质表达宿主细胞的现有技术为适用的,但仍存在对提高宿主细胞中的蛋白质表达的其他方法的需求。
技术实现要素:
[0005]
本发明的实施方式是关于具有经改良蛋白质(例如抗体)生产效率的新类型宿主细胞。新类型宿主细胞是经基因工程改造以改变一或多种基因,出乎意料地发现这些基因会影响蛋白质表达或分泌。这类基因不同于之前已知的基因,诸如靶向用于操作提高蛋白质表达的转录后调节组件。本发明中所操作的基因无关于转录、转录后调节、翻译或翻译后事件。因此,出人意料的是,遏制这些基因表达可能导致提高的蛋白质表达/分泌。
[0006]
根据本发明的实施方式,宿主细胞的基因工程改造可能涉及一或多种选自以下的基因的减弱:hdac8、dab2、凋亡蛋白酶3(caspase3)、sys1、ergic3、grasp及trim23。通过现有技术中已知的任何适合的基因工程改造技术(诸如与靶基因的rna干扰)实现基因减弱。靶向这些基因的rnai可通过将合适构建体转染至细胞中以减弱这些靶基因表达,从而产生经工程改造的宿主细胞。
[0007]
在较佳实施方式中,宿主细胞为cho细胞,且靶基因可为hdac8、dab2或凋亡蛋白酶3基因或其组合。用短发夹rna(shrna)或sirna抑制或遏制这些基因中的一个或多个会产生可支持提高的蛋白质表达和/或分泌的宿主细胞。举例而言,凋亡蛋白酶3的shrna抑制可使得宿主细胞细胞凋亡减少。因此,通过sirna或shrna,通过短期抑制或长期抑制来抑制凋亡蛋白酶3基因可提高蛋白质(例如抗体)产量。
[0008]
根据本发明的一些实施方式,hdac8、dab2或凋亡蛋白酶3的sirna或shrna抑制可产生稳定细胞系。这些稳定细胞系经过评估其转染效率、抗体表达提高、乳酸代谢、生长速率、新培养基适应性和/或长期传代(例如60代或多于60代)稳定性之后加以选择。除了能够
产生/分泌更多蛋白质(例如抗体)之外,这些稳定细胞也具有较高稳定性的特征且能够适应新培养基。因此,其适用于下游制备方法开发。
[0009]
本发明的一个方面是关于用于蛋白质表达的宿主细胞。根据本发明的一实施方式,相较于野生型细胞,该宿主细胞包含较低hdac8、dab2、凋亡蛋白酶3、sys1和/或trim23基因的表达水平。举例而言,经工程改造的宿主细胞具有明显基因减弱15%或15%以上的较低表达水平。即,相较于野生型细胞,该基因减弱的细胞可以85%水平或低于85%水平表达出特定基因。
[0010]
根据本发明的较佳实施方式,具有较低表达水平的基因为hdac8、dab2、凋亡蛋白酶3或其组合。根据基因的一些实施方式,宿主细胞为cho细胞。根据本发明的一些实施方式,转录后调节基因或细胞凋亡基因的较低表达水平是由rna干扰造成。
[0011]
通过以下描述、附图及随附的权利要求书,本发明的其他方面将变得显而易见。
附图说明
[0012]
图1显示在1c9细胞中的各种基因减弱后的蛋白质生产水平。1c9为较低igg生产细胞系(分批培养的第6天为1.28mg/l)。1c9细胞是用于判断igg分泌是否可通过各种基因的sirna抑制来增强。
[0013]
图2显示用于基因减弱的候选基因,其是如选自使用nimblegene及agilent的基因矩阵的较高及较低产量细胞的分析。
[0014]
图3显示具有靶基因减弱的各种细胞株中的蛋白质(阿瓦斯汀(avastin)及赫赛汀(herceptin))表达水平。
[0015]
图4显示适用于shrna构建体的非慢病毒载体(质粒)。
[0016]
图5显示衍生自慢病毒用于构建shrna的质粒。
[0017]
图6显示shrna的序列形式的一个实例。
[0018]
图7显示使用各种嘌呤霉素浓度下的细胞库的选择。
[0019]
图8显示用嘌呤霉素选择的各种转染物细胞的蛋白质表达水平,如图7中所描述。
[0020]
图9显示说明用于分离单个克隆经工程改造的细胞的程序的示意图。
[0021]
图10显示具有凋亡蛋白酶3减弱的前五名单克隆的蛋白质表达水平(短暂表达)。
[0022]
图11a显示前5名单克隆在长期培养(长达6周)所分析它们的特性(群体倍增时间、乳酸水平、凋亡蛋白酶3基因表达量)的结果。
[0023]
图11b显示在长期培养期间不同时间前5名单克隆的凋亡蛋白酶3减弱水平。
[0024]
图12说明前5名单克隆的长期稳定性,显示随时间变化的倍增时间及活细胞百分比(多达100代)。
[0025]
图13显示使用前3名单克隆随时间变化(第0周、第3周及第6周)的蛋白质表达水平。
[0026]
图14a显示衍生自前3名单克隆的第二代细胞的特性。
[0027]
图14b显示根据本发明的实施方式的cho细胞的转染率。
[0028]
图14c显示第二代细胞中凋亡蛋白酶3的表达水平。
[0029]
图15显示相比于第一代细胞及母细胞的第二代细胞的蛋白质表达水平。
[0030]
图16显示本发明cho细胞中所产生的赫赛汀的聚糖概况,显示主要聚糖包括g0f、
g1fa、g1fb及g2f,与市售赫赛汀的那些聚糖类似。
具体实施方式
[0031]
本发明的实施方式是关于研发用于蛋白质生产的新细胞系。这些新宿主细胞具有经改良的蛋白质(例如抗体)产量和/或分泌效率。这些细胞中所操作的基因与转录、转录后调节、翻译或翻译后事件无直接关联性,且因此,出人意料的是,遏制这些基因的表达可导致蛋白质表达/分泌提高。
[0032]
通过分析cho细胞基因组及转录组,本发明的发明人发现遏制hdac8、dab2、凋亡蛋白酶3、sys1、ergic3、grasp及trim23基因中的一个或多个可产生具有经改良的蛋白质表达和/或分泌的cho细胞。在细胞分裂期间调节染色体的结构及组织中涉及hdac8。dab2为一种适配器蛋白,其具有充当所选运送蛋白质的网格蛋白介导的内饮作用所需的网格蛋白相关分选蛋白质(clasp)的功能。负责细胞凋亡执行的半胱天冬酶的活化级联中涉及凋亡蛋白酶3。在细胞凋亡开始时,凋亡蛋白酶3会以蛋白水解方式分解聚(adp-核糖)聚合酶(parp)。sys1为位于高尔基体的整合膜蛋白质同源物且涉及高尔基体转运。trim23(含有三联基元的23)在形成自一个隔室移动至另一个的细胞内转运囊泡方面起作用。ergic3编码循环膜蛋白质,其为内质网-高尔基体中间隔室(ergic)蛋白质,其与此蛋白质家族的其他成员相互作用以提高其转换。grasp编码充当分子支架的蛋白质,其将受体(包括第1组代谢型谷氨酸受体)连接至神经元蛋白质。这些基因中无一者直接涉及转录或翻译调节。因此,遏制这些基因可导致蛋白质表达和/或分泌提高的事实是出人意料的。
[0033]
根据本发明的实施方式,分析cho染色体及转录组以选择用于工程改造的靶基因。简言之,分析cho共生物种所产生的基因芯片。在一个实施例中,分析总共四种不具有转基因的cho细胞系。其中一个细胞系为cho-s生产细胞系,而另外三个细胞系为已人工培养于实验室中的悬浮液cho细胞系。
[0034]
此外,分析具有较高或较低产率特征的三对cho细胞。在较高产率cho细胞中以较低水平表达但在较低产率cho细胞中以较高水平表达的基因为可能不利于蛋白质的高水平表达的基因。因此,这类基因可为用于rnai干扰的候选靶标以改良宿主细胞中的蛋白质表达。为了研究这类基因是否实际上会影响蛋白质表达/分泌效率,通过rna干扰降低这些靶基因的表达水平。制备用于这些基因干扰的各种rnai构建体。这些构建体瞬时转染至cho细胞中以筛选其对蛋白质表达及细胞生长及稳定性的作用。
[0035]
如图1中所示,hdac8、trim23、sys1、ergic3、dab2及grasp的基因减弱可使得经修饰细胞中的蛋白质表达和/或分泌提高。进一步地,发现hdac8及dab2基因减弱的组合在蛋白质表达改良方面产生最主要效果。具体地,减弱hdac8和dab2两个基因的细胞产生可表达1.65倍至1.8倍蛋白质。
[0036]
使用类似途径,分析两个额外cho细胞基因矩阵,一个来自agilent(santa clara,ca),而一个来自roche nimblegen(madison,wi)。较低产率细胞系中高度表达的那些基因鉴别作为干扰的靶标。通过这些分析,两个额外基因(bax和凋亡蛋白酶3)鉴别为基因减弱候选物,如图2中所示。
[0037]
基于这些靶基因,可以使用抑制这些基因功能的各种rna干扰的方法。举例而言,可构建含有靶序列(hdac8+dab2、凋亡蛋白酶3、bax、和凋亡蛋白酶3+bax)的shrna质粒。本
发明的实施方式可使用任何适合的质粒或载体。举例而言,慢病毒质粒或pcdna3.1(+)载体常用于shrna。可使用商购试剂盒,诸如来自thermo fisher scientific(waltham,ma)的block-it
tm
慢病毒rnai表达系统。
[0038]
这些质粒可在细菌中扩增。随后,将含有shrna的质粒转染至相关细胞系(如dxb11-s1)中。评定转染体的转染功效(基于质粒中的抗生素抗性,例如通过测定使用0.5-10μg/ml嘌呤霉素的杀死曲线)且分析它们基因遏制的长期效应。这些细胞系中靶基因的基因遏制可使用实时pcr分析。最终,用生物化学、细胞或分子生物学方法来分析这些细胞以研究这些基因的功能。
[0039]
除了这些基因单独的干扰之外,也探究这些靶基因的基因减弱的组合。如图3中所示,hdac8和dab2基因减弱的组合使得大部分细胞系(1c9、1g9、1g7、1e3和3c8)中的蛋白质表达水平提高1.28倍至1.88倍。相比于较高产量细胞(1e3、3c8和3g7),生产促进在较低产量细胞(1c9、1g9和1g7)中更显著。以这些细胞中所表达的不同抗体(阿瓦斯汀及赫赛汀)可见这些提高,显示产量提高将适用于蛋白质生产,一般不限于特定蛋白质。
[0040]
两个靶基因的基因减弱组合可同时产生协同作用。举例而言,hdac8及dab2的基因减弱分别在1c9细胞中产生1.28倍及1.32倍阿瓦斯汀产量,而hdac8和dab2的基因减弱组合在相同细胞中则产生1.65倍表达。类似地,hdac8及dab2的基因减弱在1g9细胞中分别产生0.78倍及1.32倍阿瓦斯汀产量,而hdac8和dab2的基因减弱组合在相同细胞中产生1.88倍表达,显示具有协同作用。
[0041]
允许在宿主细胞中瞬时且稳定的转染以及shrna表达盒的稳定递送的各种shrna载体是可用的。根据本发明的实施方式,shrna构建体转染至cho细胞中可使用任何适合的载体,包括商购载体,诸如pcdna3.1(+)(图4,可购自thermo fisher,waltham,ma)和pgfp-c-shlenti(图5,可购自origene,rockville,md)。也可使用现有技术中已知的其他适合的载体。
[0042]
尽管慢病毒载体可提供适宜方法以将shrna构建体递送及整合至细胞基因组中,在不使用慢病毒组件的情况下产生药物蛋白是较佳的。以下实施例表明来自thermo fisher(waltham,ma)的pcdna3.1(+)的用途。
[0043]
使用pcdna3.1(+)(图4)质粒作为实施例,可将具有图6中所示的结构的茎-环序列/框架插入质粒中。图6中的寡核苷酸说明典型的茎环结构构建体。用这些质粒已制备含有用于靶基因的序列的若干构建体。这些质粒的成功构造可用限制酶消化物确认以产生正确尺寸的片段。
[0044]
这些构建体是用于转染cho细胞,诸如dxb11细胞。随后评定经转染细胞的靶基因抑制。如图7中所示,可基于可选择标志物(例如嘌呤霉素抗性)选择各种经转染的细胞系(dxb11 sh-hdac8+dab2、dxb11 sh-凋亡蛋白酶3、dxb11 sh-bax-库、和dxb11 sh-bax+凋亡蛋白酶3),且随后筛选出靶基因的更佳抑制。简言之,用各种浓度的抗生素(例如嘌呤霉素)筛选这些经转染的细胞系,且用实时pcr评定靶基因的遏制。基于这些筛检,鉴别最佳候选宿主细胞。这些最佳细胞系包括例如用50μg/ml嘌呤霉素选择的dxb11sh-hdac8+dab2(hd50p)、用10μg/ml嘌呤霉素选择的dxb11 sh-凋亡蛋白酶3(c10p)、用7.5μg/ml嘌呤霉素选择的dxb11 sh-bax(b7.5p)、和用10μg/ml嘌呤霉素选择的dxb11 sh-bax+凋亡蛋白酶3(bc10p)。
[0045]
进一步评估最佳候选宿主细胞支持提高蛋白质生产的能力。用各种蛋白质,诸如seap(分泌的碱性磷酸酶)、赫赛汀及阿瓦斯汀的产量测试这些细胞。如图8中所示,大部分这些细胞系在各种培养条件下产生较多蛋白质。
[0046]
为了获得稳定细胞系,这些细胞可经连续稀释且挑取单克隆。使用用10μg/ml嘌呤霉素选择的dxb11 sh-凋亡蛋白酶3细胞(c10p)作为实施例,使用图9中所说明的程序,使用clonepix或任何其他适合的设备/方案分离单克隆。图9说明用于分离单克隆的一个方案。简言之,在细胞转染之后,扩增且筛选细胞,且随后亚克隆。细胞系可适应无血清化学成分确定的培养基中的悬浮培养物。随后,转染体的稳定细胞库可在产生较高产量稳定单克隆之前产生并加以特性分析。本领域技术人员将了解,此仅用于说明且也可使用用于实现单克隆的分离的其他程序。
[0047]
如图9中所示,这些细胞中的基因表达的遏制可使用实时pcr确认。通过这些分析,可获得具有不同靶基因(例如凋亡蛋白酶3)基因减弱百分比的细胞。随后,在适合的浓度(例如各5ml孔中3
×
105个细胞/毫升)下将这些细胞接种于6孔板中且培养适当的持续时间。测量这些细胞的活细胞密度(vcd)、群体倍增时间(pdt)及乳酸水平以评估细胞健康状态。
[0048]
随后,可用携载蛋白质基因(例如igg)的表达载体的瞬时转染,探究这些细胞中的靶基因减弱对蛋白质产量的作用。在这些细胞确认支持高水平蛋白质表达后,可通过将选择药物(例如遗传霉素(geneticin)或嘌呤霉素)添加至培养基中来选择稳定细胞库。首先滴定恰当药物浓度以使用且随后在药物的所选浓度下使细胞生长,如此仅具有所选抗性标志物的转染体是具有活力的且可以合理的速率生长。
[0049]
在已选择细胞的稳定群之后,这些细胞库可进一步经稀释且测试其稳定性。举例而言,这些稳定细胞库可进行限制性稀释并且选择出亚克隆,评定该亚克隆的稳定性。
[0050]
最终,必要时,可分离稳定转染体的单克隆以确定研究细胞库(rcb),自其中可获得主要细胞库(mcb)或工作细胞库(wcb),并以低温保存。具体言之,可基于单克隆特性(例如细胞系稳定性及蛋白质生产效率)评估单克隆且将选择最佳表达细胞系用于产生rcb。可在作为mcb低温保存之前进一步测试且表征rcb细胞。
[0051]
使用来自dxb11-sh-凋亡蛋白酶3转染体的前5名的单克隆作为实例,探究这些细胞支持蛋白质表达的效率。如图10中所示,相比于母体dxb11-j1.0细胞,所有5个克隆(ci-1b、ci-1h、cii-1h、cii-4g、和cii-3b)在经含有赫赛汀或阿瓦斯汀的载体瞬时转染后显示出提高的蛋白质表达。提高范围是1.5倍至2.4倍。这些结果显示凋亡蛋白酶3的遏制可导致较高蛋白质产量的宿主细胞。该研究结果出人意料,因为发现凋亡蛋白酶-1的遏制是在杆状病毒昆虫细胞(sf9细胞)表达系统中导致经改良的蛋白质折叠,而非提高蛋白质产量或积聚量(x.zhang等人,bmc生物技术,2018年5月2日,18(1):24;doi:10.1186/s12896-018-0434-1)。
[0052]
进一步探究这些前5名的克隆的长期状况。如图11a中所示,自第0周至第6周,这5个克隆的细胞密度不具有显著变化,与接种密度(d0、d4及d5,0.3-25
×
106个细胞/毫升)无关。细胞数目的长期稳定性显示这些细胞具有长期存活率,其可能归因于细胞凋亡的遏制。
[0053]
这些细胞的群体倍增时间(pdt)在16至19小时范围内且随时间推移未显示显著变化。在相同条件下,这些群体倍增时间与未经转染的cho细胞相当。同样,这些结果显示,凋
亡蛋白酶3经遏制的细胞具有与亲本cho细胞实质上相同的生物特性。
[0054]
在分批补料过程中,长期培养可在培养基中产生显著乳酸及氨积聚量。较高水平乳酸不利于细胞生长及产物质量。cho细胞也显示与较高乳酸产量相关的失调葡萄糖代谢,其可导致培养基酸化或非所需重量摩尔渗透浓度变化。因此,乳酸产量可用作细胞健康状态的指标。
[0055]
如图11a中所示,这些细胞培养物中的乳酸水平在6周时间段内无显著变化。乳酸水平以及乳酸/细胞数目相对较低。这些前几名克隆的较低乳酸水平暗示,这些细胞可高效地利用能量源(葡萄糖)。
[0056]
图11b显示凋亡蛋白酶3基因的表达水平。所有5个克隆具有较低凋亡蛋白酶3表达且表达水平在6周时间段内无实质性变化。这些结果显示,凋亡蛋白酶3基因的遏制是相对稳定。
[0057]
如图12中所示,这些细胞长时间(超过100代)维持近100%活力。另外,这些细胞的群体倍增时间(pdt)相对恒定。这些结果均表明,这些转染物细胞长期(例如100代或多于100代)非常稳定。
[0058]
这些细胞不仅长期稳定,而且这些细胞支持提高蛋白质表达的能力也极其稳定。如图13中所示,处cii-4g克隆外(该克隆显示在第6周表达水平有些降低),阿瓦斯汀及赫赛汀的表达水平维持在实质上相同的水平。
[0059]
为了进一步研究这些细胞的长期健康状态,通过连续稀释产生这些细胞的第二代,并挑选出如上文所描述的单克隆。也对这些第二代细胞的各种特性进行评估。
[0060]
表1
[0061][0062]
表1显示衍生自第一代细胞ci-1b的4种第二代细胞(ci-1b-d3、ci-1b-e2、ci-1b-f6、和ci-1b-g5)及衍生自第一代细胞cii-4g的2种第二代细胞(cii-4g-f6和cii-4g-g6)的分析结果。这些第二代细胞的群体倍增时间(pdt)与第一代细胞的类似,表明对于第二代细胞略微较低的生长速率。然而,差值极小。另外,乳酸水平以及每个细胞的乳酸水平在第二代细胞中也略高。
[0063]
图14a显示三种例示性第二代克隆c1-1b-d3、cii-4g-g5、和cii-4g-g6的特性。这些第二代克隆的长期稳定性可通过9周培养后几乎仍100%的存活率而得到证明。另外,这些细胞随时间推移维持一致转染效率。如图14b中所示,cho-c(即,cii-4g-g6)和ci-1b-g5细胞自0至9周维持类似的转染率(约15%)。
[0064]
如实时pcr所评定的这些第二代细胞中的凋亡蛋白酶3基因表达水平变化超过第一代细胞(图14c)。举例而言,ci-1b-d3和ci-1b-e2细胞仍显示对凋亡蛋白酶3基因具有极
好的遏制,但ci-1b-f6和cii-4g-f6相对于第一代有极小的变化。有趣的是,cii-4g-g6细胞具有凋亡蛋白酶3基因的明显经改良的遏制,而ci-1b-f6细胞则失去抑制凋亡蛋白酶3表达的能力。
[0065]
图15显示第二代细胞评估结果,支持提高的抗体(阿瓦斯汀及赫赛汀)表达。如所示,相比于对照细胞(dxb-11),这些第二代细胞显示1.21-2.4倍的这些抗体的表达水平。
[0066]
上述结果清楚地显示,凋亡蛋白酶3以及hdac8、dab2、sys1、和trim23的基因减弱可以产生出具有提高的生产能力的cho细胞。已发现各种cho细胞系,包括cho-dxb11、cho-s、cho-k1、和choc细胞的这样的提高能力。另外,各种蛋白质已在这些细胞中表达,包括针对her2、间皮素(msln)、和t细胞免疫球蛋白及粘蛋白结构域3(tim3)的抗体。一般而言,这些细胞可以约200mg/l或更高的水平产生蛋白质(抗体)。
[0067]
这些细胞中表达的蛋白质具有标准特性,包括翻译后修饰。作为一实施例,将dxb11和choc细胞中所表达的赫赛汀与商购赫赛汀进行比较且发现具有类似分子量(约145kda)。相比于市售赫赛汀/曲妥珠单抗(trastuzumab),rp-hplc(还原性及非还原性)揭示用这些细胞所产生的赫赛汀具有类似条带图谱。图16显示choc细胞中所产生的赫赛汀的n-键联聚糖概况(在pngase f释放之后),如acquity uplc beh聚糖管柱(1.7μm;waters corp.,milford,ma,usa)所分析且用乙腈和50mm甲酸铵的梯度洗脱。聚糖分析揭示,此蛋白质主要含有g0f、g1fa、g1fb、和g2f聚糖。
[0068]
上述描述清楚地展现本发明的实施方式。出乎意料地发现与蛋白质生产无直接关联的基因会影响蛋白质表达和/或分泌。根据本发明的实施方式,选择这些基因作为rnai靶标。靶向这些基因的rnai通过将恰当构建体转染至细胞中,减弱这些靶基因表达,从而工程改造宿主细胞(例如cho细胞)。
[0069]
本发明的实施方式显示,通过sirna和shrna,通过短期抑制或长期抑制,抑制所选靶基因(例如hdac8、dab2及凋亡蛋白酶3基因)可产生支持提高的蛋白质表达和/或分泌的细胞。这些结果表明,所选基因抑制宿主细胞作为用于蛋白质药物生产的新宿主具有极大潜能。尽管特定实施例是使用凋亡蛋白酶3来说明本发明的实施方式,也可针对基因减弱靶向其他基因(尤其hdac8、dab2、和hdac8+dab2)以改良蛋白质产量和/或分泌。
[0070]
用于各种程序的方法为现有技术中已知的。以下描述提供示例性程序及各种实施例以说明本发明的实施方式。本领域技术人员将了解,这些实施例仅用于说明且其他变化及修改在不脱离本发明的范围的情况下是可能的。举例而言,下文使用hdac8、dab2、bax、和凋亡蛋白酶3作为实施例以说明本发明的实施方式。然而,可在不脱离本发明的范围的情况下使用其他基因。
[0071]
细胞培养和培养基
[0072]
中国仓鼠卵巢(cho)细胞系dxb11获自哥伦比亚大学的lawrence chasin博士。在5%co2,37℃和95%湿度下,在培育箱中进行细胞培养。用于细胞培养的培养基包括hyclone和混合培养基(50%cdforticho和50%acticho)。在用锥虫蓝染色之后,使用自动细胞计数器tc10(bio-rad,美国)进行细胞计数及存活率分析。
[0073]
转染构建体包括嘌呤霉素抗性基因。基于嘌呤霉素抗性选择稳定库。一旦其变成单克隆后,就不需要选择。
[0074]
载体构建
[0075]
在此实施例中,选择hdac8、dab2、bax、和凋亡蛋白酶3作为用于rna干扰的靶基因。用于rna干扰的这些基因的所选特定序列片段显示于表2中:
[0076]
表2
[0077]
标靶靶序列seq id nohdac5'-gcatacaggatgagaagta-3'1dab25'-cagcaaagcagaagagaat-3'2bax5'-ccaaagtgcccgagctaat-3'3凋亡蛋白酶35'-cgatagaatttgagtcctt-3'4
[0078]
将这些靶序列克隆至pcdna3.1(+)载体(图5)中,以产生用于抑制这些基因的shrna质粒,从而产生经工程改造的宿主细胞。
[0079]
质粒构造如下:设计单独引物,以在5'端含有mfei限制位点及在3'端含有kpni限制位点。使用pgfp-c-dab2和pgfp-c-bax作为模板,进行pcr以使含有u6启动子及嘌呤霉素选择基因的所需片段(对应于所需rna序列)扩增。gfp-c-dab2和p-gfp-c-bax为通过将对应于所需rna序列的聚核苷酸克隆至pgfp-c-shlenti载体(图6)中由origene构建的hush shrna质粒。
[0080]
单独地设计在5'端具有ecori限制位点及在3'端具有xmai限制位点的类似引物。以类似方式,使用pgfp-hdac8和pgfp-c-凋亡蛋白酶3作为模板进行pcr以使含有u6启动子及嘌呤霉素选择基因的所需片段(对应于所需rna序列)扩增。表3显示各种引物序列:
[0081]
表3:寡核苷酸序列
[0082][0083]
pcr片段瞬时克隆至pjet1.2载体(thermo fisher)中且确认序列。分别使用限制酶mfei/kpni和ecori/xmai自暂时pjet1.2载体切割hdac8、dab2、bax、和凋亡蛋白酶3片段。
[0084]
用限制酶ecori和xmai切割pcdna3.1(+)载体。随后,分别将hdac8和凋亡蛋白酶3片段克隆至载体中以获得pcdna3.1(+)-hdac8和pcdna3.1(+)-凋亡蛋白酶3载体。
[0085]
用限制酶mfei和kpni切割pcdna3.1(+)-hdac8载体,且随后将dab2片段构建至切割载体中以获得pcdna3.1(+)-dab2-hdac8载体。
[0086]
另外,pcdna3.1(+)和pcdna3.1(+)-凋亡蛋白酶3载体用限制酶mfei和kpni切割。随后,使bax片段与切割载体连接以获得pcdna3.1(+)-bax和pcdna3.1(+)-bax-凋亡蛋白酶3。构建这些载体后,通过限制酶消化确认恰当构建以确认恰当片段(即,合适尺寸)构建至载体中。
[0087]
cho细胞转染
[0088]
在6孔板中培养dxb11细胞。各孔在3ml含有8mm glutamax
tm
(thermo fischer)的hyclone
tm hycell
tm cho培养基(ge healthcare)中具有3
×
106个细胞。
[0089]
可使用现有技术中已知的任何适合的试剂,诸如亲脂性试剂freestyle max(thermo fischer)进行含有shrna构建体的载体(如pcdna3.1)的转染。举例而言,将rnai/
shrna载体及转染剂freestylemax
tm
(thermo fischer)分别添加至optipro
tm sfm(thermo fischer)中以制备如表4中所示的载体溶液。这些溶液静置5分钟,之后将其添加至转染试剂中且充分混合。使所得溶液静置20分钟,之后转染至细胞中。转染之后3天评估细胞。
[0090]
表4
[0091]
rnai载体转染
[0092][0093][0094]
shrna载体转染
[0095][0096]
蛋白质表达
[0097]
对于测试cho细胞中的蛋白质/抗体产生,抗体/蛋白质表达构建体可来自市售来源或基于现有技术中已知的程序制备且经转染至用于抗体或蛋白质的短暂表达的测试cho细胞中。培养经转染的cho细胞适当的持续时间(例如3天)以产生抗体。
[0098]
可使用任何适合的方法评定蛋白质表达水平。举例而言,greatescape
tm
化学荧光试剂盒获自clontech。用ddh2o以1:5稀释5x稀释缓冲液来制备1x稀释缓冲液。
[0099]
为了评估蛋白质表达水平,将25μl细胞培养基自转染细胞或模拟转染细胞移转至96孔微量滴定板中。必要时,板可密封且冷冻在-20℃下以便将来分析。在96孔微量滴定板中将75μl 1x稀释缓冲液添加至各样品中。用粘着铝箔或规则96孔盖子密封该板且使用加热块或水浴在65℃下培育经稀释的样品30分钟。
[0100]
在冰上冷却样品2-3分钟,随后平衡至室温。将100μl seap受体溶液添加至各样品中。在读取之前在室温下培育30分钟。使用96孔酶标仪(例如)检测且记录化学荧光信号。
[0101]
使用实时pcr的基因减弱分析
[0102]
靶基因表达水平可用qpcr评估。简言之,来自细胞的rna使用rna纯化试剂(来自qiagen)提取且使用nanodrop 2000定量。使用以下条件进行qpcr反应(表5):
[0103]
表5
[0104][0105]
使用stepone实时pcr机,用以下方案进行qpcr(表6):
[0106]
表6
[0107][0108]
为了评估凋亡基因(凋亡蛋白酶3和bax)及其他基因(例如hdac8和dab2)的表达水平,使用以下引物(表7):
[0109]
表7
[0110][0111][0112]
即使本发明的实施方式已用有限数目的实施例加以说明,本领域技术人员仍应理
解,其他修饰及变化在不脱离本发明的范围的情况下是可能的。因此,本发明的保护范围应仅受所附权利要求书限制。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips