
 商标分类
商标分类  商标转让
商标转让 
具有空间电荷转移效应的荧光材料及其应用的制作方法
 2021-02-02 07:02:00|
2021-02-02 07:02:00| 345|
345| 起点商标网
起点商标网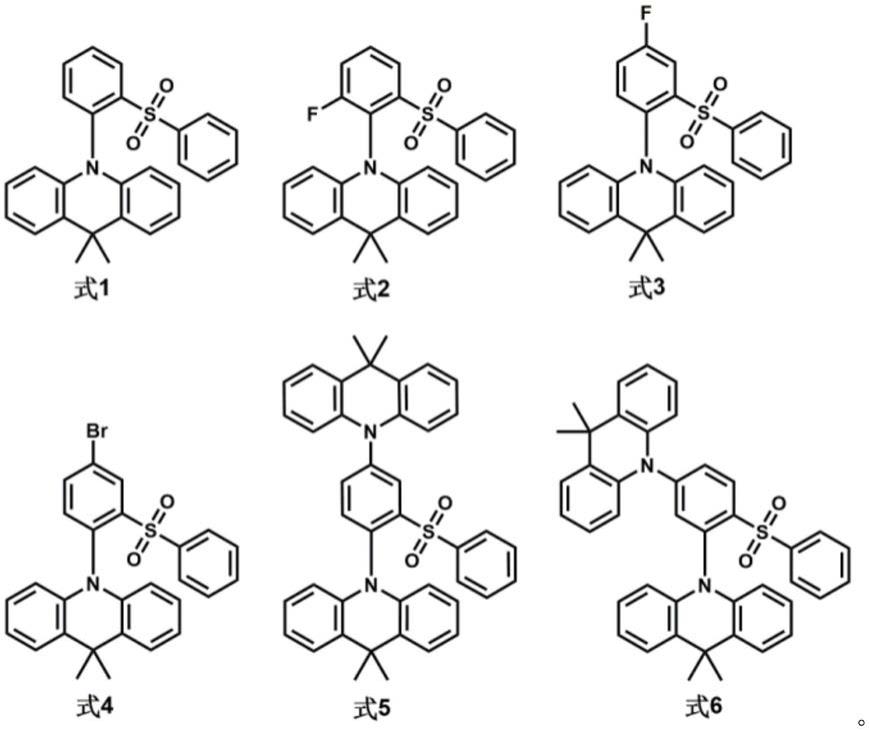
[0001]
本发明属于有机电致发光材料技术领域,主要涉及一类具有空间电荷转移效应的新型荧光材料,及其在发光器件中的应用。
背景技术:
[0002]
tadf过程是由于最低激发单线态(s1)和最低激发三线态(t1)之间的能级差(δe
st
)足够小,三线态激子受热后通过反系间窜越(risc)跃迁到单线态,单线态激子再辐射跃迁回基态而发光。因此,基于tadf材料的器件内量子效率理论上能达到100%。
[0003]
而高效的电子耦合是构建光电功能π系统的关键。通常,π电子的离域必须符合由共价键(通常为σ键)构成的框架,代表经典的键间共轭。然而,通过空间共轭可以提供一种替代方案,实现与紧密堆积的π系统而非共价键的空间电子作用,从而实现多维能量和电荷传输。贯穿空间共轭是π电子离域的最重要特性之一,可导致紧密面对面重叠的芳环之间发生非共价环间相互作用,由于其非共价结构和π电子的空间离域而具有更大的灵活性和可能性。
[0004]
1949年第一次对对环芳烷(pcp)的报导象征着贯穿空间共轭研究的开始。由于它们令人着迷的光致发光和电荷传输特性,许多科研人员开始利用空间共轭分子来构建具有多维能量和电荷转移能力的各种功能材料,其在光电材料和器件应用中具有很高的前景。
技术实现要素:
[0005]
为了获得更高效的tadf材料,本发明通过合理的分子设计,利用空间共轭构筑了系列具有空间电荷转移效应的新型高效tadf材料。二甲基吖啶是一种强给电子单元,常用作tadf材料的给体,而二苯基砜是一种具有多个修饰位点的常用弱受体单元,将二者有机结合可以实现一定的分子内扭曲,实现tadf性能。另一方面,本发明在二苯基砜的邻位引入电子给体单元,形成分子内空间共轭,这更加有利于电子传输,从而获得高效的tadf材料;利用不同的基团对二苯基砜受体的不同位点进行修饰,可以进一步改善材料的发光性能。
[0006]
为了实现上述技术目的,本发明以二苯基砜及其衍生物为受体单元,并在二苯基砜单元的邻位引入给体单元,提供了一类具有空间电荷转移效应的新型高效tadf材料,该材料具有式1~式6结构:
[0007][0008]
上述结构的荧光材料具有热活性延迟荧光、聚集诱导发光、力致发光和室温磷光性质。
[0009]
本发明的另一个目的是提供tadf材料的应用,将荧光材料作为发光层掺杂剂,制备发光器件,可以获得发光性能优异的发光二极管。
[0010]
相对现有技术,本发明的技术方案带来的有益效果在于:
[0011]
1、在二苯基砜单元的邻位引入给体单元,有利于形成分子内空间共轭,使材料具有空间电荷转移效应,这有利于减少单三线态能隙差和提高材料的发光效率,从而获得高效的tadf材料;
[0012]
2、这类材料的空间电荷转移效应使材料同时具有多功能发光效应:同时具有热活化延迟荧光、聚集诱导发光、力致发光及室温磷光性能。这是首次报道的通过空间电荷转移诱导的多功能发光材料。
[0013]
3、将这类材料用于有机电致发光二极管的发光层,通过溶液加工和真空蒸镀工艺,都可获得高效率发光的电致发光器件。
附图说明
[0014]
图1为本发明实施例1制得的化合物1~6的热失重曲线图。
[0015]
图2为本发明实施例1制得的化合物1~6的循环伏安曲线图。
[0016]
图3为本发明实施例1制得的化合物1~6在甲苯溶液中的紫外可见光吸收以及光致发光光谱图。
[0017]
图4为本发明实施例1制得的化合物1在甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺,四种溶剂中的光致发光光谱图。
[0018]
图5为本发明实施例1制得的化合物2在甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺,四种溶剂中的光致发光光谱图。
[0019]
图6为本发明实施例1制得的化合物3在甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺,四种溶剂中的光致发光光谱图。
[0020]
图7为本发明实施例1制得的化合物4在甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺,四种溶剂中的光致发光光谱图。
[0021]
图8为本发明实施例1制得的化合物5在甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺,四种溶剂中的光致发光光谱图。
[0022]
图9为本发明实施例1制得的化合物6在甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺,四种溶剂中的光致发光光谱图。
[0023]
图10为本发明实施例1制得的化合物1~6在四氢呋喃/水溶液体系中的光致发光光谱图。
[0024]
图11为本发明实施例1制得的化合物1的荧光(fl)、磷光(ph)和力致发光(ml)光谱图。
[0025]
图12为本发明实施例1制得的化合物2的荧光、磷光光谱图。
[0026]
图13为本发明实施例1制得的化合物3的荧光、磷光和力致发光光谱图。
[0027]
图14为本发明实施例1制得的化合物1~3的延迟寿命曲线图。
具体实施方式
[0028]
下面结合实施例对本发明做进一步描述,但不限于此。
[0029]
实施例1
[0030]
基于二甲基吖啶,二苯基砜及其衍生物的新型tadf材料合成的具体步骤如下:
[0031][0032]
化合物sm1的合成
[0033]
于100ml单口瓶中依次加入苯硫酚(4.6ml,45.05mmol)、1-氟-2-碘苯(5.2ml,45.05mmol)、碘化亚铜(857mg,4.51mmol)、邻菲罗啉(811mg,4.51mmol)、碳酸钾(18.65g,135.15mmol)、50ml甲苯,混合物在氮气保护下加热至120℃回流反应24小时。冷却至室温,减压旋蒸除去甲苯,二氯甲烷萃取(3
×
30ml),水洗三次,无水硫酸镁干燥,过滤,收集滤液,旋干溶剂,以石油醚为洗脱剂经柱层析分离得到无色油状液体7.8g,产率85%。1h nmr(500mhz,cdcl3)δ7.55
–
7.46(m,3h),7.31(dt,j=13.3,4.5hz,4h),7.26
–
7.18(m,2h).
[0034]
化合物sm2的合成
[0035]
于200ml单口瓶中加入sm1(7.8g,38.28mmol)和50ml冰醋酸,搅拌下滴加50ml的过氧化氢,加热至100℃回流反应12小时。冷却至室温,将混合物倒入100ml水中,析出白色固体,过滤,收集固体,烘干,用乙醇重结晶,得到目标化合物5.8g,为白色固体,产率65%。1h nmr(500mhz,dmso)δ8.08(td,j=7.7,1.7hz,1h),7.96(d,j=8.4hz,2h),7.85
–
7.73(m,2h),7.67(dd,j=10.7,4.9hz,2h),7.51(td,j=7.8,0.9hz,1h),7.46
–
7.38(m,1h).
13
c nmr(126mhz,cdcl3)δ160.25,158.20,140.89,136.04,135.97,133.70,129.76,129.14,128.17,124.62,117.39,117.23.
[0036]
化合物sm3的合成
[0037]
将1-氟-2-碘苯换成1,6-2氟-2碘苯,合成步骤与化合物sm1的合成相同,以石油醚为洗脱剂经柱层析分离得到无色油状液体4.5g,产率97%。1h nmr(500mhz,cdcl3)δ7.42
–
7.37(m,2h),7.37
–
7.30(m,3h),7.10
–
7.03(m,1h),6.98(tdd,j=8.0,4.8,1.4hz,1h),6.93(ddt,j=7.8,6.0,1.6hz,1h).
[0038]
化合物sm4的合成
[0039]
将sm1换成sm3,合成步骤与化合物sm2的合成相同,用乙醇重结晶,得到白色固体3.9g,产率77%。1h nmr(500mhz,dmso)δ8.00(d,j=7.9hz,2h),7.95
–
7.83(m,2h),7.83
–
7.77(m,1h),7.70(dd,j=10.8,4.9hz,2h),7.54(td,j=8.4,3.3hz,1h).
13
c nmr(126mhz,cdcl3)δ151.87,151.76,149.85,149.75,148.78,148.66,146.71,146.60,140.35,134.08,131.70,131.62,129.33,128.26,128.25,124.65,124.61,124.56,124.37,124.34,123.12,122.98.
[0040]
化合物sm5的合成
[0041]
将1-氟-2-碘苯换成1,4-2氟-2碘苯,合成步骤与化合物sm1的合成相同,以石油醚为洗脱剂经柱层析分离得到无色油状液体7.7g,产率99%。1h nmr(500mhz,dmso)δ7.45
–
7.29(m,6h),7.22
–
7.13(m,1h),6.90(ddd,j=8.6,5.8,3.0hz,1h).
[0042]
化合物sm6的合成
[0043]
将sm1换成sm5,合成步骤与化合物sm2的合成相同,用乙醇重结晶,得到白色固体6.8g,产率77%。1h nmr(500mhz,dmso)δ8.00(d,j=8.4hz,2h),7.89(ddd,j=7.6,5.4,3.3hz,1h),7.81
–
7.75(m,1h),7.74
–
7.64(m,3h),7.52(td,j=9.4,4.0hz,1h).
13
c nmr(126mhz,cdcl3)δ159.01,158.99,157.03,157.01,156.20,156.18,154.18,154.16,140.18,134.07,129.28,128.34,128.33,122.65,122.59,122.46,122.39,118.93,118.87,118.74,118.68,116.59,116.37.
[0044]
化合物sm7的合成
[0045]
将1-氟-2-碘苯换成1氟-2-碘-4溴苯,合成步骤与化合物sm1的合成相同,以石油醚为洗脱剂经柱层析分离得到无色油状液体9.2g,产率98%。1h nmr(500mhz,cdcl3)δ7.46
–
7.29(m,6h),7.22(dd,j=6.5,2.5hz,1h),6.97(t,j=8.8hz,1h).
[0046]
化合物sm8的合成
[0047]
将sm1换成sm7,合成步骤与化合物sm2的合成相同,用乙醇重结晶,得到白色固体7.9g,产率78%。1h nmr(500mhz,cdcl3)δ8.23(dd,j=6.2,2.5hz,1h),8.06
–
7.97(m,2h),7.70
–
7.62(m,2h),7.59
–
7.52(m,2h),7.01(t,j=9.0hz,1h).
13
c nmr(126mhz,cdcl3)δ159.20,157.15,140.16,138.75,138.69,134.13,132.23,129.32,128.32,128.31,119.30,119.12,117.15,117.12.
[0048]
化合物sm9的合成
[0049]
将1-氟-2-碘苯换成1,5-2氟-2碘苯,合成步骤与化合物sm1的合成相同,以石油醚为洗脱剂经柱层析分离得到无色油状液体8.7g,产率94%。1h nmr(500mhz,cdcl3)δ7.36(td,j=8.4,6.3hz,1h),7.31
–
7.27(m,3h),7.27
–
7.20(m,2h),6.92
–
6.83(m,2h).
[0050]
化合物sm10的合成
[0051]
将sm1换成sm9,合成步骤与化合物sm2的合成相同,用乙醇重结晶,得到白色固体
8.3g,产率82%。1h nmr(500mhz,cdcl3)δ8.14(td,j=8.4,6.2hz,1h),8.00(d,j=8.3hz,2h),7.64(t,j=7.4hz,1h),7.55(t,j=7.8hz,2h),7.09
–
7.00(m,1h),6.92
–
6.82(m,1h).
13
c nmr(126mhz,cdcl3)δ167.57,167.48,165.52,165.42,161.12,161.01,159.05,158.95,140.67,133.88,131.70,131.61,129.32,129.29,129.26,129.23,129.21,129.20,129.18,128.06,128.05,126.03,126.00,125.92,125.89,112.30,112.27,112.13,112.10,106.08,105.88,105.68.
[0052]
化合物1的合成
[0053]
于100ml双口瓶中加入化合物sm2(1g,4.23mmol)、9,10-二氢-9,9-二甲基吖啶(1.03g,5.08mmol)和氢化钠(338mg,8.46mmol),氮气保护下在体系中加入30ml新蒸的四氢呋喃,加热至70℃回流反应24h。冷却至室温,二氯甲烷萃取(3
×
30ml),水洗三次,无水硫酸镁干燥,过滤,收集滤液,旋干溶剂,以石油醚:二氯甲烷(3:1,v/v)为洗脱剂经柱层析分离得到白色固体640mg,产率36%。1h nmr(500mhz,cdcl3)δ8.48(dd,j=8.0,1.5hz,1h),7.99(td,j=7.6,1.6hz,1h),7.91(td,j=7.8,1.2hz,1h),7.50(dd,j=8.4,1.1hz,2h),7.45(dd,j=7.8,1.4hz,2h),7.41
–
7.34(m,2h),7.17(dd,j=8.2,7.6hz,2h),6.85
–
6.74(m,2h),6.67
–
6.58(m,2h),5.41(dd,j=8.3,0.9hz,2h),1.78(s,3h),1.66(s,3h).
13
c nmr(126mhz,cdcl3)δ142.38,141.23,139.53,139.16,136.62,134.49,132.77,131.44,129.48,129.34,128.34,128.31,126.22,126.16,120.70,114.65,36.36,35.54,32.48.
[0054]
化合物2的合成
[0055]
将sm2换成sm4,合成步骤与化合物1的合成相同,以石油醚:二氯甲烷(3:1,v/v)为洗脱剂经柱层析分离得到淡黄色固体810mg,产率47%。1h nmr(500mhz,cdcl3)δ8.36(dt,j=8.1,1.2hz,1h),7.74(td,j=8.2,4.9hz,1h),7.66
–
7.49(m,3h),7.40(dd,j=7.8,1.4hz,2h),7.27
–
7.23(m,1h),7.14
–
7.01(m,2h),6.89
–
6.79(m,2h),6.66(ddd,j=8.5,7.3,1.5hz,2h),5.59(d,j=8.3hz,2h),1.81(s,3h),1.76(s,3h).
13
c nmr(126mhz,cdcl3)δ162.08,160.02,144.27,138.67,138.41,133.12,130.76,130.69,129.49,128.47,128.44,128.26,128.14,126.46,123.46,123.30,121.21,113.35,36.07,35.52,32.71.
19
f nmr(471mhz,cdcl3)δ-114.52.
[0056]
化合物3的合成
[0057]
将sm2换成sm6,合成步骤与化合物1的合成相同,以石油醚:二氯甲烷(3:1,v/v)为洗脱剂经柱层析分离得到淡黄色固体400mg,产率23%。1h nmr(500mhz,dmso)δ8.27(dd,j=8.3,3.1hz,1h),7.92
–
7.83(m,1h),7.55(dd,j=8.3,1.0hz,2h),7.50
–
7.43(m,3h),7.40(t,j=7.5hz,1h),7.18(dd,j=8.1,7.7hz,2h),6.84
–
6.74(m,2h),6.68
–
6.57(m,2h),5.45(dd,j=8.2,0.9hz,2h),1.78(s,3h),1.65(s,3h).
13
c nmr(126mhz,cdcl3)δ162.89,160.87,144.46,144.41,139.43,138.44,137.05,137.02,136.48,136.41,133.14,129.44,128.47,126.35,126.24,123.87,123.69,120.90,118.72,118.52,114.51,36.34,35.52,32.53.
19
f nmr(471mhz,dmso)δ-108.66.
[0058]
化合物4的合成
[0059]
将sm2换成sm8,合成步骤与化合物1的合成相同,以石油醚:二氯甲烷(3:1,v/v)为洗脱剂经柱层析分离得到黄色固体630mg,产率39%。1h nmr(500mhz,dmso)δ8.58(d,j=2.3hz,1h),8.19(dd,j=8.3,2.4hz,1h),7.56(d,j=7.4hz,2h),7.45(dd,j=7.8,1.1hz,
2h),7.39(t,j=7.5hz,1h),7.34(d,j=8.4hz,1h),7.17(t,j=7.9hz,2h),6.84
–
6.76(m,2h),6.67
–
6.59(m,2h),5.45(d,j=7.8hz,2h),1.77(s,3h),1.65(s,3h).
13
c nmr(126mhz,cdcl3)δ144.19,140.22,139.83,139.18,138.45,136.20,134.26,133.17,129.40,128.48,128.47,126.38,126.27,123.21,120.98,114.56,36.38,35.51,32.53.
[0060]
化合物5的合成
[0061]
于100ml单口瓶中依次加入化合物4(600mg,1.19mmol)、9,10-二氢-9,9-二甲基吖啶(299mg,1.43mmol)、叔丁醇钠(572mg,5.95mmol)、三叔丁基膦四氟硼酸盐(35mg,0.12mmol)、醋酸钯(13mg,0.06mmol)和30ml甲苯,混合物在氮气保护下加热至110℃回流反应48h。冷却至室温,旋干溶剂,二氯甲烷萃取(3
×
30ml),水洗三次,无水硫酸镁干燥,过滤,收集滤液,旋干溶剂,以石油醚:二氯甲烷(3:1,v/v)为洗脱剂经柱层析分离得到黄色固体560mg,产率74%。1h nmr(500mhz,cdcl3)δ8.58(d,j=2.4hz,1h),7.83(dd,j=8.2,2.5hz,1h),7.69
–
7.59(m,2h),7.58
–
7.50(m,3h),7.43(dd,j=7.8,1.3hz,2h),7.28(t,j=7.4hz,1h),7.20
–
7.00(m,6h),6.93
–
6.84(m,2h),6.75(dd,j=11.2,4.2hz,2h),6.39(d,j=8.1hz,2h),5.68(d,j=8.2hz,2h),1.86(s,3h),1.79(s,3h),1.74(s,6h).
13
c nmr(126mhz,cdcl3)δ145.16,142.51,140.47,140.35,139.70,139.39,138.81,137.09,134.19,133.07,131.12,129.54,128.53,128.39,126.66,126.47,126.39,125.59,121.67,121.00,114.43,114.11,36.41,36.19,35.60,32.51,30.93.
[0062]
化合物6的合成
[0063]
将sm2换成sm10,合成步骤与化合物1的合成相同,以石油醚:二氯甲烷(3:1,v/v)为洗脱剂经柱层析分离得到黄色固体400mg,产率16%。1h nmr(500mhz,dmso)δ8.72(d,j=8.6hz,1h),7.95(dd,j=8.6,2.1hz,1h),7.66
–
7.59(m,2h),7.48(dd,j=7.8,1.3hz,2h),7.45
–
7.36(m,4h),7.22(t,j=7.9hz,2h),7.08
–
7.02(m,2h),6.95(td,j=7.7,1.0hz,2h),6.82
–
6.75(m,2h),6.73
–
6.66(m,2h),6.39(dd,j=8.2,0.8hz,2h),5.65(dd,j=8.2,0.8hz,2h),1.68(d,j=10.9hz,6h),1.55(s,6h).
13
c nmr(126mhz,cdcl3)δ149.99,143.90,140.80,140.03,139.25,139.07,134.49,133.70,133.00,132.78,129.62,129.40,128.51,126.55,126.47,126.33,125.42,122.23,120.93,115.75,114.61,36.39,36.36,35.52,32.83,30.44.
[0064]
实施例2
[0065]
在35~500℃范围内,以20℃每分钟的速率升温,测得化合物1~6的热失重曲线。由图1可知,化合物1~6分解5%的温度分别为277℃、275℃、262℃、275℃、353℃、371℃。
[0066]
实施例3
[0067]
用循环伏安法测得化合物1~6的氧化还原电势,如图2所示,根据公式和带边吸收计算得出:化合物1的homo能级为-5.31ev,lumo能级为-2.29ev;化合物2的homo能级为-5.41ev,lumo能级为-2.57ev;化合物3的homo能级为-5.32ev,lumo能级为-2.44ev;化合物4的homo能级为-5.35ev,lumo能级为-2.55ev;化合物5的homo能级为-5.29ev,lumo能级为-2.39ev;化合物6的homo能级为-5.30ev,lumo能级为-2.46ev。
[0068]
实施例4
[0069]
将实施例1中的化合物1~6溶解在甲苯中配成10-5
mol/l的溶液,测试其溶液的紫外可见吸收和光致发光光谱。由图3可知,化合物在溶液中的紫外可见吸收光谱大致有两种
吸收峰:短波长(283nm)处的吸收峰主要归属于分子的π-π*的跃迁吸收;长波长(330~420nm)的吸收峰归属于分子内给体单元到受体单元的电荷转移(ict)跃迁吸收峰。另外,如图3所示,在300nm波长激发下,化合物1的最大发射峰为509nm,化合物2的最大发射峰为543nm,化合物3的最大发射峰为533nm,化合物4的最大发射峰为538nm,化合物5的最大发射峰为532nm,化合物6的最大发射峰为523nm。
[0070]
实施例4
[0071]
实施例1中的化合物1~6在不同溶液中的光致发光性能测试。将化合物1~6分别溶解在甲苯、二氯甲烷、四氢呋喃、n,n-二甲基甲酰胺溶剂中,测试其在不同溶剂中的光致发光光谱,如图4~9所示。由图可知,在光激发下,化合物的发射波长随着溶剂极性的增加呈现出一定的红移,说明这类化合物具有较强的分子内电荷转移。
[0072]
实施例5
[0073]
将化合物1~6溶解在四氢呋喃中,加水配制成不同水含量的10-5
mol/l的溶液,用300nm波长激发测试其发射光谱。如图10所示,随着水占比例的增大,发射强度先不断降低,达到80%、90%后突然升高,表明化合物1~6都有聚集诱导荧光增强特性。
[0074]
实施例6
[0075]
测试了化合物1~3固体粉末的荧光、磷光和力致发光光谱,如图11~13所示:在粉末状态下化合物1的荧光发射峰为467nm,磷光发射峰为504nm,力致发光发射峰为481nm;化合物2的荧光发射峰为497nm,磷光发射峰为525nm,力致发光发射峰为481nm;化合物3的荧光发射峰为502、525nm,磷光发射峰为509、532nm,力致发光发射峰为513nm。
[0076]
实施例7
[0077]
在氮气氛围下测试了化合物1~3固体粉末的荧光寿命,如图14所示,经计算得出这3个化合物的延迟寿命分别为2.14μs、3.03μs、2.66μs,均为长寿命。在固体粉末状态下,测试化合物1-3的荧光量子效率分别为:96.77%、88.84%和99.70%。
[0078]
尽管结合了优选实施例对本发明进行了说明,但本发明并不局限于上述实施例,应当理解所附权利要求概括了本发明的范围。在本发明构思的指导下,本领域的技术人员应当意识到,对本发明的各实施例方案所进行的一定的改变,都将被本发明的权利要求书的精神和范围所覆盖。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips