
 商标分类
商标分类  商标转让
商标转让 
含吲唑结构的咪唑类衍生物及其制备方法和应用与流程
 2021-02-02 07:02:56|
2021-02-02 07:02:56| 371|
371| 起点商标网
起点商标网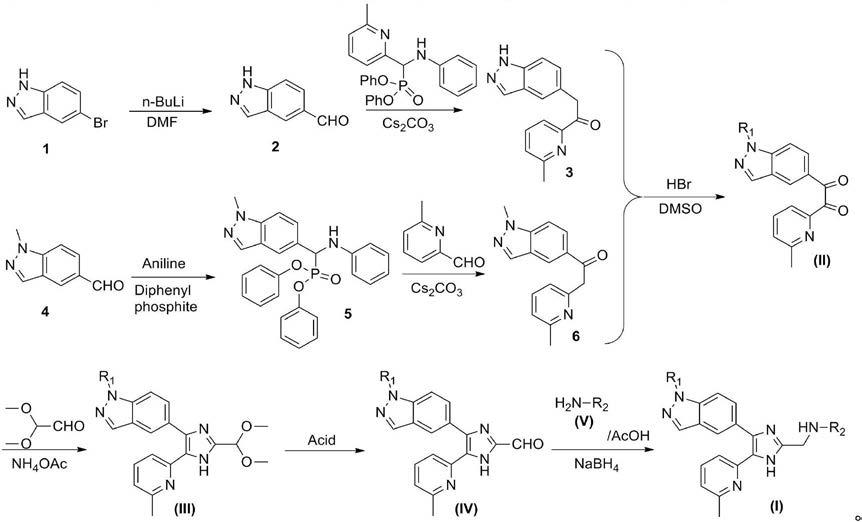
yoon,dh nam.targeting the epithelial to mesenchymal transition in glioblastoma:the emerging role of met signaling.onco targets ther.2014,20(7):1933-1944.)。emt是一种生物学过程,其中上皮细胞失去其细胞极性和细胞间粘附,从而获得间充质细胞的迁移和侵袭性质。emt过程的特征在于上皮标志物(e-cadherin)表达降低以及间质标志物(vimentin、n-cadherin等)表达增加。尽管关于胶质母细胞瘤的emt存在争议,但在神经上皮细胞中,越来越多的证据已经证实了胶质母细胞瘤中emt样过程的存在。激活胶质母细胞瘤emt样程序已被证明可促进恶性进展,涉及体外和体内迁移和侵袭。tgf-β是目前研究最为深入的emt诱导因子,尤其是由肿瘤细胞或基质成纤维细胞分泌的细胞因子,其主要作用有炎症细胞趋化、促进细胞增殖分化和迁移、影响血管形成、控制细胞外基质合成与降解等。tgf-β通过smad信号通路、non-smad信号通路调控肿瘤emt,同时诱导emt转录因子及相关的转录调节因子的表达,如snail、slug和twist等。slug又称为snail2,是重要的emt转录因子,增强肿瘤细胞的迁移和侵袭能力,并参与多种emt相关分子的调节。slug使形态分化的基因表达降低,而使tgf-β通路标志物如富含半胱氨酸的分泌型酸性蛋白通过维持slug的高表达促进emt相关肿瘤的侵袭性,并且使tgf-βr ii启动子区域组蛋白的乙酰化增强。emt过程的两种调节模式:一种涉及转录过程如slug或snail1对细胞粘连分子的抑制,另一种与tgf-β及其它转导途径导致的细胞迁移有关。slug诱导emt表型至少部分通过tgf-β信号分子起作用。emt的发生涉及多个信号传导途径,如tgf-β/smad通路、wnt/β-catenin通路、磷脂酰肌醇3激酶(pi3k/akt)通路、src通路、il-6/stat3通路、整合素通路、notch通路、hedgehog通路以及nf-κb通路等。各通路之间关系复杂,相互影响,协同发挥作用。这些信号通路在转录后水平激活emt转录因子(emt-tfs),从而调节emt过程,促进肿瘤的侵袭和转移。肿瘤侵袭及转移是指肿瘤细胞从原发部位脱落,通过血液、淋巴及直接浸润等方式在不连续的靶部位长出具有相同性质的肿瘤的过程。肿瘤的侵袭与转移的潜力取决于肿瘤细胞以及促肿瘤细胞相关的内环境之间的相互作用。emt一方面可以改变肿瘤细胞的特性使肿瘤细胞间连接分子表达缺失,极性丧失黏附能力减弱、运动性提高,使肿瘤本身的侵袭和转移以及血管形成的微环境,增强肿瘤侵袭和转移能力。因此,emt在肿瘤的侵袭和转移中发挥重要作用。此外,emt还会使一些参与细胞外基质、基底膜降解和破坏的溶解酶高表达,破坏正常的组织学屏障,使肿瘤细胞更易从原发部位脱落而发生侵袭和转移。emt不仅与肿瘤的侵袭、转移、复发和治疗抵抗有关,而且还能刺激肿瘤细胞获得干细胞特征,emt、cscs和耐药构成了致命性“三组合”或癌症的“邪恶轴心”,成为肿瘤难以治愈的根本原因。因此,靶向抑制emt,可能阻止肿瘤细胞的侵袭和转移、清除cscs并克服耐药性,起到“一箭三雕”的作用,已成为备受重视的肿瘤治疗新策略。emt已在上皮性肿瘤中的作用已进行了广泛研究,在乳腺癌、结直肠癌、胰腺癌、甲状腺癌和肺癌等均证明,emt是进展性上皮性肿瘤获得侵袭、转移和耐药能力的重要细胞学基础。与上皮癌细胞不同,由于其神经上皮起源,这种emt过程尚未在恶性胶质母细胞瘤中被广泛接受。然而,有证据表明原发性胶质母细胞瘤和其干细胞系表达间充质干细胞的细胞和分子特征。之前的研究还发现,对治疗有反应且复发前时间较长的胶质母细胞瘤患者肿
瘤样本中的间充质特征水平较低。此外,与间充质特性相关的分子或该过程的激活剂如zeb2,slug/snail2和twist的过表达显着促进胶质母细胞瘤的迁移,侵袭进展。因此,它表明emt或间充质(特性)可能是癌症治疗的理想靶点,不仅在上皮癌细胞中,而且在胶质母细胞瘤中。鉴于emt在恶性肿瘤中的作用,现在需要更有效的小分子emt抑制剂。alk5抑制剂能直接抑制emt过程。这些化合物能抑制多种emt信号传导途径。因此,开发具有强效的emt抑制活性的药物,必将在临床上有着良好的应用前景。
技术实现要素:
本发明的目的在于提供含吲唑结构的咪唑类衍生物及其制备方法和应用。本发明所述含吲唑结构的咪唑类衍生物具有转化生长因子β1受体激酶(alk5)抑制活性,能够抑制tgf-β诱导的emt样过程,对恶性胶质瘤具有潜在抗癌作用,可用于治疗涉及上皮间质转化(emt)样过程的人神经胶质瘤。本发明提供了含吲唑结构的咪唑类衍生物,所述咪唑类衍生物为结构式如式(i)所示的含吲唑结构的咪唑类化合物及其在药学上可接受的盐或水合物:其中:r1为h或ch3;r2为无取代或被取代的苯基。优选的是,所述r2包括苯基、邻氟苯基、间氟苯基、对氟苯基、邻甲基苯基、间甲基苯基或对甲基苯基。优选的是,所述咪唑类衍生物包括:n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-氟苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-氟苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-氟苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-甲基苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-甲基苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-甲基苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-氟苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-氟苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-氟苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-甲基苯
胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-甲基苯胺;或n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-甲基苯胺及其药学上可接受的盐或水合物。本发明还提供了上述技术方案所述咪唑类衍生物的制备方法,包括以下步骤:1)将式a所示化合物与有机金属锂试剂或格式试剂混合进行离子交换反应,得到锂或格式试剂取代的化合物,将所述锂或格式试剂取代的化合物与无水dmf进行加成反应,得到1h-吲唑-5-甲醛;所述x为卤族元素;2)将1h-吲唑-5-甲醛与二苯基(6-甲基吡啶-2-基)-苯胺甲基磷酸酯混合,在碱性条件下进行缩合反应,加入酸进行水解反应,得到2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮;3)将1-甲基-1h-吲唑-5-甲醛和苯胺进行缩合反应生成亚胺化合物,将所述亚胺化合物与亚磷酸二苯酯进行加成反应,得到二苯基((1-甲基-1h-吲唑-5-基)(苯氨基)甲基)-膦酸盐;4)将6-甲基吡啶-2-甲醛和二苯基((1-甲基-1h-吲唑-5-基)(苯氨基)甲基)-膦酸盐在碱性条件下进行缩合反应,加入酸进行水解反应,得到1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮;5)将2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮或1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮溶于二甲基亚砜,加入hbr,得到α-溴代的单酮化合物,60~80℃进行氧化反应1~2h,得到1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮;6)将1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮溶于醇类溶剂,依次加入乙酸铵和乙二醛二甲基缩醛,45~55℃进行环合反应2~8h,得到5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑;7)将5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑与酸混合,65~75℃进行缩醛的水解反应2~6h,得到4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛;8)将4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛与苯胺类化合物混合,在乙酸的参与下,75℃进行缩合反应2h,脱水生成亚胺化合物,将所述亚胺化合物与还原剂混合,进行还原反应,得到结构式如式(i)所示的含吲唑结构的咪唑类化合物;所述苯胺类化合物的结构式为h2n-r2;
所述步骤1)和3)没有时间先后顺序的限定。本发明还提供了一种alk5激酶抑制剂,所述抑制剂包括上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制tgf-β诱导的上皮间质转化样过程的药物中的应用。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制肿瘤细胞侵袭能力的药物中的应用。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制肿瘤入侵和/或转移的药物中的应用。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备治疗人神经胶质瘤的药物中的应用。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制癌症的药物中的应用。本发明提供了含吲唑结构的咪唑类衍生物。本发明含吲唑结构的咪唑类衍生物不但可以抑制emt转录因子,而且还可以调控emt间充质标志性蛋白的表达;同时,亦能抑制细胞转移和侵袭能力。使其作为一类新型小分子药物应用在抑制gbm肿瘤上皮间质转化领域。此外,本发明结果为alk5抑制剂发展为治疗恶性胶质瘤的药物提供了一个线索。本发明所述含吲唑结构的咪唑类衍生物具有转化生长因子β1受体激酶(alk5)抑制活性,能够抑制tgf-β诱导的emt样过程,调控转录因子slug的表达,下调与emt相关的人神经胶质瘤u87细胞间充质形态、转移和入侵,对恶性胶质瘤具有潜在抗癌作用,可用于治疗涉及上皮间质转化(emt)样过程的人神经胶质瘤。试验结果表明,本发明所述化合物的活性远高于阳性对照化合物ly-2157299,其中化合物i-1-1和i-1-3的抑制活性最强,其中,j-1090(i-1-3)剂量性抑制tgf-β诱导emt转录因子slug及间充质标志性蛋白(n-cadherin、vimentin),导致下调细胞转移和侵袭能力。具体的,j-1090以剂量依赖性方式抑制tgf-β诱导的间充质形态,其效果优于阳性对照药ly-2157299;以剂量依赖性方式抑制tgf-β诱导u87细胞核内slug表达,其效果优于阳性对照药ly-2157299;以剂量依赖性方式抑制tgf-β诱导的emt间充质标志物的表达,n-cadherin、vimentin表达下调,其效果优于阳性对照药ly-2157299;抑制tgf-β诱导的emt间充质标志物mrna水平;几乎完全阻断tgf-β诱导u87细胞核内slug表达;几乎完全阻断tgf-β诱导u87细胞n-cadherin表达;抑制tgf-β增加的肿瘤细胞转移能力;抑制tgf-β增加的肿瘤细胞侵袭能力。
附图说明
图1为本发明提供的光学显微镜下观察不同处理对u87细胞形态的影响;图2为本发明提供的western blot法观察不同处理对tgf-β诱导u87细胞核内slug表达的影响;图3为本发明提供的western blot法观察不同处理对tgf-β诱导的emt间充质标志物、n-cadherin、vimentin表达的影响;图4为本发明提供的pcr法观察不同处理对tgf-β诱导的emt间充质标志物mrna水平的影响;图5为本发明提供的免疫荧光法观察不同处理对tgf-β诱导u87细胞核内slug表达的影响;图6为本发明提供的免疫荧光法观察不同处理对tgf-β诱导u87细胞n-cadherin表达的影响;图7为本发明提供的划痕法观察不同处理对tgf-β增加的肿瘤细胞转移能力的影响;图8为本发明提供的入侵法观察不同处理对tgf-β增加的肿瘤细胞侵袭能力的影响。
具体实施方式
本发明提供了含吲唑结构的咪唑类衍生物,所述咪唑类衍生物为结构式如式(i)所示的含吲唑结构的咪唑类化合物及其在药学上可接受的盐或水合物:其中:r1为h或ch3;r2为无取代或被取代的苯基。本发明r1为h时,吲唑的-nh可以与alk5的关键蛋白有结合,显示较强药理作用;当r1为ch3时,甲基成为空间位阻,不能与alk5的关键蛋白有结合,显示较弱药理作用。在本发明中,术语“被取代”是指特定苯环上的任意一个氢原子被取代基取代,只要特定原子的价态是正常的并且取代后的化合物是稳定的即可。本发明所述含吲唑结构的咪唑类衍生物具有转化生长因子β1受体激酶(alk5)抑制活性,能够抑制tgf-β诱导的emt样过程,调控转录因子slug的表达,下调与emt相关的人神经胶质瘤u87细胞间充质形态、转移和入侵,对恶性胶质瘤(包括人神经胶质瘤)具有潜在抗癌作用。在本发明中,所述r2包括苯基、邻氟苯基、间氟苯基、对氟苯基、邻甲基苯基、间甲基苯基或对甲基苯基。本发明上述取代基的选取相比于其它取代基显示较强的alk5抑制活性。在本发明中,所述咪唑类衍生物包括:n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-氟苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-氟苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-氟苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-甲基苯胺;n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-甲基苯胺;
n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-甲基苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-氟苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-氟苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-氟苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-甲基苯胺;n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-甲基苯胺;或n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-甲基苯胺及其药学上可接受的盐或水合物。本发明上述具体衍生物具有明确的靶点和作用机制;不但可以抑制emt转录因子,而且还可以调控emt间充质标志性蛋白的表达,同时,亦能抑制肿瘤细胞转和侵袭能力。这些衍生物克服了传统的抗癌药物的毒性大、作用机制不明确的问题。本发明还提供了上述技术方案所述咪唑类衍生物的制备方法,包括以下步骤:1)将式a所示化合物与有机金属锂试剂或格式试剂混合进行离子交换反应,得到锂或格式试剂取代的化合物,将所述锂或格式试剂取代的化合物与无水dmf进行加成反应,得到1h-吲唑-5-甲醛;所述x为卤族元素;2)将1h-吲唑-5-甲醛与二苯基(6-甲基吡啶-2-基)-苯胺甲基磷酸酯混合,在碱性条件下进行缩合反应,加入酸进行水解反应,得到2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮;3)将1-甲基-1h-吲唑-5-甲醛和苯胺进行缩合反应生成亚胺化合物,将所述亚胺化合物与亚磷酸二苯酯进行加成反应,得到二苯基((1-甲基-1h-吲唑-5-基)(苯氨基)甲基)-膦酸盐;4)将6-甲基吡啶-2-甲醛和二苯基((1-甲基-1h-吲唑-5-基)(苯氨基)甲基)-膦酸盐在碱性条件下进行缩合反应,加入酸进行水解反应,得到1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮;5)将2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮或1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮溶于二甲基亚砜,加入hbr,得到α-溴代的单酮化合物,60~80℃进行氧化反应1~2h,得到1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮;6)将1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮溶于醇类溶剂,依次加入乙酸铵和乙二醛二甲
基缩醛,45~55℃进行环合反应2~8h,得到5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑;7)将5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑与酸混合,65~75℃进行缩醛的水解反应2~6h,得到4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛;8)将4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛与苯胺类化合物混合,在乙酸的参与下,75℃进行缩合反应2h,脱水生成亚胺化合物,将所述亚胺化合物与还原剂混合,进行还原反应,得到结构式如式(i)所示的含吲唑结构的咪唑类化合物;所述苯胺类化合物的结构式为h2n-r2;所述步骤1)和3)没有时间先后顺序的限定。本发明所述咪唑类衍生物的制备中,化学反应关系如下式所示:本发明将式a所示化合物与有机金属锂试剂或格式试剂混合进行离子交换反应,得到锂或格式试剂取代的化合物,将所述锂或格式试剂取代的化合物与无水dmf进行加成反应,得到1h-吲唑-5-甲醛;所述x为卤族元素,在本发明中,所述卤族元素优选为氯(cl)、溴(br)、碘(i),更优选为溴;
在本发明中,所述式a所示化合物与有机金属锂试剂或格式试剂的摩尔比优选为1:(2~5),更优选为1:2.5;所述有机金属锂试剂优选包括正丁基锂。在本发明中,当所述式a所示化合物中的x为溴时,所述化合物为5-溴-1氢-吲唑(式1所示化合物1)。当使用5-溴-1氢-吲唑(化合物1)和正丁基锂进行反应时,本发明优选在5-溴-1氢-吲唑(化合物1)中滴加正丁基锂进行离子交换反应。本发明优选在所述离子交换反应过程中进行搅拌,之后再加入无水dmf进行加成反应。在本发明中,与有机金属锂试剂或格式试剂反应的式a所示化合物优选以式a所示化合物溶液的形式提供,所述式a所示化合物溶液的溶剂优选为四氢呋喃;所述式a所示化合物溶液中式a所示化合物与溶剂的用量比优选为1mmol:(3~5)ml,更优选为1mmol:4ml。在本发明中,所述式a所示化合物溶液的制备优选为:采用溶剂溶解所述式a所示化合物;对所得溶解液置于-45~-55℃,优选-50℃环境下冷却20min,冷却的作用是使反应液的温度充分冷却下来,使所述离子交换反应在低温下进行,避免发生副反应。在本发明实施例中,本发明优选将取6.00g 5-溴-1氢-吲唑(化合物1,30.46mmol)于250ml圆底烧瓶中,向其中加入四氢呋喃120ml使原料溶解;再在溶解液中滴加48ml正丁基锂(76.15mmmol)。本发明在所述加成反应后,优选进行搅拌、萃取、洗涤、干燥、过滤、减压蒸干和层析的操作,实现化合物的纯化,得到1h-吲唑-5-甲醛。在本发明中,所述搅拌的时间优选为25~60min,更优选为30min。搅拌后,本发明优选将加成反应后得到的混合物经水淬后,置于室温(20~25℃)条件下,本发明所述水淬的目的是使反应液中未反应的有机金属锂或格式试剂失活,进而终止反应,水淬能为后续萃取扫除障碍。在本发明中,所述萃取优选使用乙酸乙酯进行,实现目标化合物的分离;萃取的次数优选为3次,多次萃取后,优选合并有机相。在本发明中,所述洗涤优选为使用饱和食盐水溶液对有机相进行洗涤。本发明所述干燥优选使用脱水剂,如无水硫酸钠,干燥有机相中的水分。本发明对所述过滤和减压蒸干的操作没有特殊限定,采用本领域技术人员熟知的化合物合成用常规过滤和减压蒸干方法条件即可。本发明所述过滤的目的为去除有机相中的已吸水的脱水剂。在本发明中,所述层析优选为使用硅胶柱进行层析,所述硅胶柱层析优选使用石油醚/乙酸乙酯=4∶1作为洗脱溶液。本发明得到的1h-吲唑-5-甲醛(化学反应关系式中化合物2所示)的结构式如式2所示:得到1h-吲唑-5-甲醛(化合物2)后,本发明将1h-吲唑-5-甲醛(化合物2)与二苯基(6-甲基吡啶-2-基)-苯胺甲基磷酸酯混合,在碱性条件下进行缩合反应,加入酸进行水解反应,得到2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮(化合物3)。本发明对所述碱性条件的提供没有特殊限定,使用本领域技术人员熟知的碱即可,不能为强碱。在本发明中,所述碱性条件优选使用碳酸铯、碳酸钠或碳酸钾提供,更优选使用碳酸铯。本发明碱性条件的提供可以使二苯基(6-甲基吡啶-2-基)-苯胺甲基磷酸酯脱氢产生碳负离子,属于酸碱中和反应,碱的加入能够助二苯基(6-甲基吡啶-2-基)-苯胺甲基磷酸酯和1h-吲唑-5-甲醛的缩合
反应。本发明所述缩合反应后能够得到亚胺化合物,本发明所述酸的添加能够中和混合溶液中的碱,促进上述生成的亚胺化合物的水解。本发明将1h-吲唑-5-甲醛(化合物2)与二苯基(6-甲基吡啶-2-基)-苯胺甲基磷酸酯混合,在碱性条件下进行缩合反应。在本发明中,当使用碳酸铯提供碱性条件时,所述1h-吲唑-5-甲醛(化合物2)、二苯基(6-甲基吡啶-2-基)-苯胺甲基磷酸酯和碳酸铯的摩尔比优选为1∶(1.2~1.5)∶(1.2~1.5)更优选为1∶1.3∶1.3;所述缩合反应的温度优选为室温(20~25℃),时间优选为10~14h,更优选为12h,溶液由无色变为淡黄色,澄清变浑浊。所述混合前,本发明优选将化合物2溶解,所述溶解用溶剂优选为四氢呋喃与异丙醇的混合溶剂,所述混合溶剂中四氢呋喃和异丙醇的体积比优选为(3~5)∶1,更优选为4∶1。在本发明实施例中,本发明优选取5.20g化合物2(35.58mmol)于250ml圆底烧瓶中,向其中加入四氢呋喃和异丙醇的4∶1混合溶剂(100ml)搅拌均匀。以5.20g化合物2为基准,本发明优选添加二苯基(6-甲基吡啶-2基)-苯胺基甲基磷酸酯(20g,46.25mmol)和碳酸铯(15g,46.25mmol)。缩合反应后,本发明加入酸进行水解反应。在本发明中,水解反应的温度优选为室温(20~25℃),时间优选为30~90min,更优选为1h。在本发明中,所述酸优选包括hcl、hbr、h3po4或柠檬酸等,更优选为hcl,最优选为1n hcl。当所述酸为1n hcl时,以5.20g化合物2为基准,所述1n hcl的用量优选为140~160ml,更优选为150ml。加入酸后,溶液由浑浊变澄清。水解反应后,本发明优选加入甲基叔丁基醚,搅拌,分离水和甲基叔丁基醚层,水层在冰水浴中调ph至中性,用乙酸乙酯萃取,得到有机相,洗涤、干燥、过滤,减压蒸干、层析后,得到2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮(化合物3)。本发明加入甲基叔丁基醚分离混合物中易溶于有机溶剂且不溶于水的杂质,在本发明中,以5.20g化合物2为基准,所述甲基叔丁基醚的加入量优选为70~80ml,更优选为75ml。本发明加入甲基叔丁基醚后搅拌的时间优选为25~35min,更优选为30min;加入甲基叔丁基醚进行搅拌后产生分层,本发明分离水和甲基叔丁基醚层,本发明优选使用分液漏斗分离水和甲基叔丁基醚层,分离后,本发明取水层进行后续操作(化合物3的分子中具有吡啶结构,这些结构在酸性条件下易成盐,溶解到水里),水层优选在冰水浴中调ph至中性,本发明优选用饱和nahco3溶液调ph至中性,ph值的调节能够使得溶于水的化合物3游离出来。本发明冰水浴的操作能够避免中和反应放热导致温度升高,避免产生副反应。在本发明中,所述萃取优选使用乙酸乙酯进行,萃取的次数优选为3次,多次萃取后优选合并有机相,本发明用乙酸乙酯萃取(乙酸乙酯的选取能够把化合物3溶于乙酸乙酯层里,且乙酸乙酯的比重小于水,因此分层后,乙酸乙酯层在上层,操作方便)。本发明优选使用饱和食盐水对得到的有机相进行洗涤,优选使用无水硫酸钠进行干燥,去除有机相中的水分,过滤的目的是要去除有机相中的已吸水的脱水剂。本发明对所述过滤和减压蒸干的操作没有特殊限定,采用本领域技术人员熟知的常规过滤和减压蒸干方法即可。本发明所述层析优选使用石油醚/乙酸乙酯=5∶1作为洗脱溶液进行层析。本发明得到的化合物3的结构式(化学反应关系式中化合物3所示)如式3所示:
吲唑-5-基)(苯氨基)甲基)-膦酸盐(化合物5)、碳酸铯的摩尔比优选为1∶(1.2~1.5)∶(1.2~1.5),更优选为1∶1.3∶1.3。在本发明具体实施例中,优选取2.6g 6-甲基吡啶-2-甲醛(21.54mmol)与150ml混合溶剂混合,搅拌均匀,再优选加入9.13g化合物5(28.00mmol)和9.13g碳酸铯(28.00mmol)。缩合反应后,本发明加入酸进行水解反应。本发明所述水解反应优选进行搅拌,所述水解反应的温度优选为室温(20~25℃),时间优选为0.5~1.5h,更优选为1h,溶液由浑浊变澄清。在本发明中,所述酸优选包括hcl、hbr、h3po4或柠檬酸等,更优选为hcl,最优选为1n hcl。当所述酸为1n hcl时,以2.6g 6-甲基吡啶-2-甲醛(21.54mmol)为基准,所述1n hcl的用量优选为80~100ml,更优选为90ml。进行水解反应后,本发明优选加入甲基叔丁基醚搅拌,分离水和甲基叔丁基醚层,水层在冰水浴中调ph至中性,用乙酸乙酯萃取,得到有机相,洗涤、干燥、过滤、减压蒸干、层析后,得到1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮(化合物6)。本发明优选加入甲基叔丁基醚搅拌,分离混合物中易溶于有机溶剂的杂质。在本发明中,2.6g 6-甲基吡啶-2-甲醛(21.54mmol)为基准,所述甲基叔丁基醚的加入量优选为40~50ml。在本发明中,加入甲基叔丁基醚后搅拌的时间优选为25~35min,更优选为30min。在本发明实施例中,以添加2.6g 6-甲基吡啶-2-甲醛(21.54mmol)为基准,甲基叔丁基醚的量优选为45ml。加入甲基叔丁基醚后,会产生分层,本发明分离水和甲基叔丁基醚层。在本发明中,所述分离优选使用分液漏斗进行。本发明优选取水层(得到的化合物6含吡啶结构,在酸性条件下易成盐,溶解到水里)在冰水浴中调ph至中性,本发明优选用饱和nahco3溶液调ph至中性,将溶于水中的化合物6游离出来。中和反应放热,本发明进行冰水浴能够避免反应溶液温度过高,避免产生副产物。在本发明中,所述萃取的次数优选为3次,所述萃取优选使用乙酸乙酯进行,多次萃取后,合并有机相。本发明合并的有机相优选用饱和食盐水洗涤。本发明优选加无水硫酸钠干燥。本发明对所述过滤和减压蒸干的操作没有特殊限定,采用本领域技术人员熟知的常规有机相的过滤和减压蒸干操作即可。本发明优选经硅胶柱层析(石油醚/乙酸乙酯=2∶1作为洗脱溶液),得固体化合物6。本发明得到的化合物6的结构式(化学反应关系式中化合物6所示)如式6所示:得到2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮(化合物3)或1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮(化合物6)后,本发明将2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮(化合物3)或1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮(化合物6)溶于二甲基亚砜,加入hbr,得到α-溴代的单酮化合物,60~80℃进行氧化反应1~2h,得到1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮。本发明将2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮(化合物3)或1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮(化合物6)溶于二甲基亚砜,加入hbr,得到α-溴代的单酮化合物。在本发明中,所述2-(1h-吲唑-5-基)-1-(6-甲基吡啶-2-基)乙酮(化合物3)
或1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮(化合物6)的物质的量与二甲基亚砜的体积比优选为1mmol∶(2~4)ml,更优选为1mmol∶3ml。在本发明中,所述二甲基亚砜既是溶剂,又是反应物,为氧化反应提供所需的氧原子。在本发明具体实施例中,本发明优选取19.90mmol的化合物3或化合物6溶于60ml二甲基亚砜中。本发明优选在搅拌条件下缓慢滴加hbr,所述hbr优选为质量百分含量为40%的hbr水溶液,来源优选为常规市售。以19.90mmol的化合物3或化合物6为基准,本发明优选滴加质量百分含量为40%的hbr水溶液17ml,即79.60mmol。得到α-溴代的单酮化合物后,本发明60~80℃进行氧化反应1~2h。在本发明中,所述氧化反应的温度更优选为70℃,所述氧化反应的时间更优选为1.5h。本发明所述氧化反应能够把上述生成的α-溴代的单酮化合物变成目标二酮化合物(1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮)。本发明在所述氧化反应后,冷却至0℃,调ph至中性,萃取,得到有机相,洗涤、干燥、过滤、减压蒸干、层析,得到1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮,即化合物ii。本发明通过冷却至0℃减少或消除后续中和反应产生的热量造成的副反应。本发明调ph至中性,能够使目标二酮化合物从水中游离出来,便于萃取,本发明优选用饱和nahco3溶液调ph至中性。本发明所述萃取优选使用二氯甲烷作为溶剂,也可使用其它溶剂,如乙酸乙酯。在本发明中,所述萃取的次数优选为3次,多次萃取后优选合并有机相。得到有机相后,本发明优选进行洗涤、干燥、过滤、减压蒸干、层析。本发明优选先水洗合并的有机相,所述水洗的次数优选为2次;水洗后,本发明优选再用饱和食盐水反复洗涤3次。本发明优选使用无水硫酸钠进行干燥。本发明对所述过滤和减压蒸干的操作没有特殊限定,采用本领域技术人员熟知的有机相常规过滤和减压蒸干操作即可。本发明优选使用硅胶柱层析(石油醚/乙酸乙酯=4∶1作为洗脱溶剂),得到固体化合物ii-1和化合物ii-2。化合物ii-1为1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮,结构式如式ii-1所示:化合物ii-2为1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮,结构式如式ii-2所示:得到化合物ii(化学反应关系式中化合物ii所示)即1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮(化合物ii-1)或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮(化合物ii-2)后,本发明将1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮(化合物ii-1)或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮(化合物ii-2)溶于醇类溶剂,依次加入乙酸铵和乙二醛二甲基缩醛,45~55℃进行环
合反应2~6h,得到5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑,即化合物iii。在本发明中,所述环合反应中,化合物ii的物质的量与醇类溶剂的体积比优选为1mmol∶(4~6)ml,更优选为1mmol∶5.3ml。在本发明中,所述醇类溶剂优选包括正醇类,如甲醇、乙醇、丙醇、丁醇等,更优选为甲醇。本发明优选在搅拌下向其中依次加入乙酸铵、乙二醛二甲基缩醛,乙酰胺易溶于醇类溶剂,先加入乙酰胺利于后续的化合物ii与乙二醛二甲基缩醛发生反应。在本发明中,依次加入乙酸铵和乙二醛二甲基缩醛,优选用甲基叔丁醚作为溶剂对乙酸铵和乙二醛二甲基缩醛进行溶解。在本发明中,所述环合反应的温度优选为50℃,时间优选为2h。本发明所述环合反应能够生成咪唑环,50℃条件下反应速度快,2h就能反应结束,上述温度和时间的限定能够保证所述环合反应顺利进行。在本发明中,所述化合物ii、乙酸铵的物质的量比优选为1∶(8~11),更优选为1∶9.9。本发明优选使用质量百分含量为55~65%,更优选为60%的乙二醛二甲基缩醛水溶液。在本发明中,所述化合物ii与乙二醛二甲基缩醛的物质的量比优选为1∶(1.4~1.5),更优选为1∶1.47。在本发明具体实施例中,本发明具体取9.05mmol的化合物ii溶于48ml的甲醇,本发明优选在搅拌的条件下依次加入乙酸铵89.48mmol,60%的乙二醛二甲基缩醛优选2.33ml,即13.3mmol,当使用甲基叔丁基醚作为溶剂时,甲基-叔丁基醚的添加量优选为48ml。环合反应后,本发明优选进行冷却,调ph至中性,萃取,得到有机相,洗涤、干燥、过滤、减压蒸干、层析,得到5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑,即化合物iii。本发明所述冷却优选冷却至室温(20~25℃),本发明所述冷却能够减少或消除后续中和反应后产生的热量造成的副反应,本发明优选冷却至室温(20~25℃)。本发明所述环合反应的条件是偏酸性,因此后处理时优选通过调ph至中性来减少产物的损失。本发明优选用饱和nahco3溶液调ph至中性。调ph至中性后,本发明进行萃取,本发明所述萃取优选使用极性有机溶剂进行,如二氯甲烷、乙酸乙酯。本发明所述萃取的次数优选为3次,多次萃取后,本发明优选合并有机相。萃取后,得到有机相,本发明进行洗涤、干燥、过滤、减压蒸干、层析。本发明优选使用饱和食盐水反复洗涤。本发明优选使用无水硫酸钠进行干燥。本发明对所述过滤和减压蒸干的操作没有特殊限定,采用常规操作即可。本发明优选使用硅胶柱层析,在本发明中,所述层析优选使用二氯甲烷和甲醇的混合溶剂(体积比20∶1)作为洗脱溶剂(或流动相),得到固体化合物iii-1和iii-2。本发明所述化合物iii-1为5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑,结构式为式iii-1所示:化合物iii-2为5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑,结构式如式iii-2所示:
得到化合物iii(化学反应关系式中化合物iii所示)即5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑后,本发明将5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑与酸混合,65~75℃进行缩醛的水解反应2~6h,得到4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛,即化合物iv。本发明酸的加入能够生成溶于水的盐溶液。本发明所述温度和时间的限定,能够在较短的时间内完成缩醛的水解反应,经济,简便,收率高。在本发明中,所述酸优选包括hcl、hbr、h3po4或柠檬酸等,更优选为hcl,最优选为1n hcl。在本发明中,当所述酸为1n hcl时,所述化合物iii优选与1n的hcl进行混合,所述化合物iii的物质的量和1n的hcl的体积比优选为1mmol∶(2~4)ml,更优选为1mmol∶3ml。在本发明实施例中,优选取化合物iii 5.74mmol与18ml 1n的hcl混合。在本发明中,所述缩醛的水解反应的温度优选为70℃,时间优选为4h。在本发明中,所述水解反应后,优选冷却至0℃,调ph至中性,抽滤,洗涤,干燥,得到4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛,即化合物iv。本发明冷却至0℃能够减少或消除后续中和反应后产生的热量造成的副反应,冷却后,本发明优选调ph至中性,使产物从水中游离出来,本发明优选用饱和nahco3溶液调ph至中性。之后,本发明抽滤,洗涤,干燥。调节ph后,生成的固体本发明优选用布氏漏斗抽滤,取滤饼,用水洗涤滤饼。本发明优选在减压干燥箱中干燥得化合物iv-1和iv-2。所述化合物iv-1为4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛,结构式如式iv-1所示:化合物iv-2为4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛,结构式如式iv-2所示。得到化合物iv(化学反应关系式中化合物iv所示)即4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛后,本发明将4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛与苯胺类化合物混合,在乙酸的参与下,75℃进行缩合反应2h,脱水生成亚胺化合物,将所述亚胺化合物与还原剂混合,
进行还原反应,得到结构式如式(i)所示的含吲唑结构的咪唑类化合物;所述苯胺类化合物的结构式为h2n-r2。本发明将化合物iv与苯胺类化合物混合,在乙酸的参与下,75℃进行缩合反应2h,脱水生成亚胺化合物。在本发明中,所述化合物iv与苯胺类化合物的物质的量比为1∶(1~3),更优选为1∶1.5。在本发明中,所述化合物iv与乙酸的物质的量比为1∶(0.5~1.2),更优选为1∶1。本发明化合物iv在反应前优选溶于无水溶剂,如无水四氢呋喃、无水1,2-二氯乙烷或无水甲苯。在本发明中,所述化合物iv的物质的量与无水溶剂的体积比为1mmol∶(20~30)ml,更优选为1mmol∶24ml。本发明优选在搅拌下加入乙酸和苯胺类化合物,上述物质混合后,常温下不会发生反应。本发明75℃,2h的限定,可以实现缩合反应的顺利进行,脱水形成亚胺化合物。本发明实施例中,优选取化合物iv 0.33mmol溶于8ml无水四氢呋喃中,搅拌下优选依次加入0.02g乙酸(0.33mmol)和0.50mmol苯胺类化合物。得到亚胺化合物后,将所述亚胺化合物与还原剂混合,进行还原反应。本发明在缩合反应后,还原反应前,优选冷却至0℃,再加甲醇,本发明甲醇添加的作用为溶解后续添加的还原剂。在本发明实施例中,以化合物iv0.33mmol为基准,所述甲醇的加入量优选为3ml。本发明加入甲醇后,优选在反应液中分次加入还原剂,在本发明中,所述还原剂优选包括硼氢化钠。在本发明实施例中,以化合物iv 0.33mmol为基准,所述硼氢化钠的加入量(分次加入的总量)优选为0.06g,即1.32mmol。本发明分次加入能够避免还原反应过于剧烈,避免反应时放出氢气。加入还原剂后,本发明升温搅拌3h,促使还原反应彻底完成。在本发明中,所述升温优选为升温至20~25℃。之后,本发明优选加水,产生猝灭反应(加水使反应液中未反应完的还原剂(硼氢化钠)失活,进而防止副反应的产生),停止反应。停止反应后,本发明调ph至中性,萃取,得到有机相,洗涤、干燥、过滤、减压蒸干、层析。本发明调ph至中性,能提高产物收率,本发明优选用1n hcl调ph至中性。本发明优选使用ch2cl2萃取,萃取的次数优选为3次,多次萃取后合并有机相。本发明萃取能够使终产物从水中分离出来,得到有机相。得到有机相后,本发明洗涤、干燥、过滤、减压蒸干、层析,得到结构式如式(i)所示的含吲唑结构的咪唑类化合物;所述苯胺类化合物的结构式为h2n-r2。本发明优选用饱和食盐水洗涤所述合并的有机相,优选加入无水硫酸钠进行干燥。本发明对所述过滤和减压蒸干的操作没有特殊限定,采用常规操作即可。本发明优选使用硅胶柱层析,本发明所述柱层析使用的洗脱溶剂(或流动相)优选为二氯甲烷和甲醇的混合溶剂(体积比20:1~10:1)。本发明还提供了一种alk5激酶抑制剂,所述抑制剂包括上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制tgf-β诱导的上皮间质转化样过程的药物中的应用。本发明所述咪唑类衍生物或抑制剂通过抑制tgf-β诱导的上皮间质转化样过程,进而抑制tgf-β诱导emt转录因子slug及间充质标志性蛋白(n-cadherin、vimentin),导致下调细胞转移和侵袭能力。具体的,本发明所述咪唑类衍生物或抑制剂能够以剂量依赖性方式抑制tgf-β诱导的间充质形态,其效果优于阳性对照药ly-2157299;本发明所述咪唑类衍生物或抑制剂能够以剂量依赖性方式抑制tgf-β诱导u87细胞核内slug表达,其效果优于阳性对照药ly-2157299;且能够以剂量依赖性方式抑制tgf-β诱导的emt
间充质标志物的表达,n-cadherin、vimentin表达下调,其效果优于阳性对照药ly-2157299;抑制tgf-β诱导的emt间充质标志物mrna水平;能够几乎完全阻断tgf-β诱导u87细胞核内slug表达;和几乎完全阻断tgf-β诱导u87细胞n-cadherin表达。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制肿瘤细胞侵袭能力的药物中的应用。本发明所述咪唑类衍生物或抑制剂能够抑制tgf-β增加的肿瘤细胞侵袭能力。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制肿瘤入侵和/或转移的药物中的应用。本发明所述咪唑类衍生物或抑制剂能够抑制tgf-β增加的肿瘤细胞转移能力。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备治疗人神经胶质瘤的药物中的应用。本发明还提供了上述技术方案所述咪唑类衍生物或上述技术方案所述制备方法制备得到的咪唑类衍生物或上述技术方案所述抑制剂在制备抑制癌症的药物中的应用。在本发明中,所述癌症包括由alk5受体介导的癌症。下面结合具体实施例对本发明所述的含吲唑结构的咪唑类衍生物及其制备方法和应用做进一步详细的介绍,本发明的技术方案包括但不限于以下实施例。本发明实施例中以下述物质作为对照:ly-2157299(g giannelli,e villa,m lahn.transforming growth factor-βas a therapeutic target in hepatocellular carcinoma.cancer res.2014,74(7):1890-1894;rj kovacs,g maldonado,a azaro,et al.cardiac safety of tgf-βreceptor i kinase inhibitor ly2157299 monohydrate in cancer patients in a first-in-human dose study.cardiovasc.toxicol.2015,15(4):309-323.)是由美国利来公司(eli lilly and company)研发的一种alk5抑制剂,目前在临床三期,用于骨髓增生异常综合症和实体瘤治疗,其商品名为galunisertib。实施例1化合物2的制备取6.00g 5-溴-1氢-吲唑(化合物1,30.46mmol)于250ml圆底烧瓶中,向其中加入四氢呋喃120ml使原料溶解。将溶液置于-50℃环境下冷却20min后,在此温度下缓慢滴加正丁基锂(48ml,76.15mmol),并搅拌2h。在该温度下加入无水dmf(4.7ml,60.92mmol),持续搅拌30min。反应混合物经水淬后,置于室温。反应溶液用乙酸乙酯萃取3次,合并有机相。合并的有机相用饱和食盐水溶液洗涤后,加无水硫酸钠干燥,过滤,减压蒸干,经硅胶柱层析(石油醚/乙酸乙酯=4∶1),得固体化合物2。化合物21h-吲唑-5-甲醛黄色固体,收率:58%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ10.54(br s,1h,nh),10.07(s,1h),8.32(s,1h),8.27(s,1h),7.99(d,1h,j=9.0hz),7.62(d,1h,j=9.0hz)。由此可知,所得化合物的结构式为:
hcl(90ml),溶液由浑浊变澄清,室温下搅拌反应1h,再加入甲基叔丁基醚(45ml),搅拌反应30min。用分液漏斗分离水和甲基叔丁基醚层,水层在冰水浴中用饱和nahco3溶液调ph至中性。反应液用乙酸乙酯萃取3次,合并有机相。合并的有机相用饱和食盐水洗涤后,加无水硫酸钠干燥,过滤,减压蒸干。粗品经硅胶柱层析(石油醚/乙酸乙酯=2∶1),得固体化合物6。化合物61-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮黄色固体,收率:23%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)(keto form,71%)δ8.57(s,1h),8.13(d,2h,j=9.0hz),7.53(t,1h,j=7.5hz),7.41(d,1h,j=9.0hz),7.12(d,1h,j=6.0hz),7.03(d,1h,j=6.0hz),4.53(s,2h),4.09(s,3h),2.55(s,3h);1h nmr(300mhz,cdcl3)(enol form,29%)δ8.27(s,1h),8.03(s,1h),7.90(d,1h,j=9.0hz),7.0(t,1h,j=7.5hz),7.41(d,1h,j=9.0hz),6.88(d,1h,j=9.0hz),6.76(d,1h,j=6.0hz),6.08(s,1h),4.06(s,3h),2.54(s,3h)。由此可知,所得化合物的结构式为:实施例5式ii化合物的制备以1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮(ii-1)和1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮(ii-2)为例介绍式ii化合物的合成通法。将化合物2-(1h-吲达唑-5-基)-1-(6-甲基吡啶-2-基)乙酮或1-(1-甲基-1氢-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙酮(19.90mmol)溶于二甲基亚砜(dmso,60ml)中,搅拌下向其中缓慢滴加40%hbr(17ml,79.60mmol)后,将反应液加热至70℃反应1.5h。反应结束后,将反应液冷却至0℃,用饱和nahco3溶液调ph至中性。反应液用二氯甲烷萃取3次,合并有机相。合并的有机相水洗2次,再用饱和食盐水反复洗涤3次,加入无水硫酸钠干燥,过滤,减压蒸干。粗品经硅胶柱层析(石油醚/乙酸乙酯=4∶1),得固体化合物ii-1和ii-2。化合物ii-1 1-(1h-吲唑-5-基)-2(6-甲基吡啶-2-基)乙烷-1,2-二酮浅黄色固体,收率:44%;进行核磁共振检测,检测结果为:1h nmr(300mhz,dmso-d6)δ13.61(s,1h),8.31(d,2h,j=9.0hz),8.06-8.01(m,2h),7.91(d,1h,j=9.0hz),7.71(d,1h,j=9.0hz),7.63(dd,1h,j=6.0,3.0hz),2.39(s,3h)。由此可知,所得化合物的结构式为:化合物ii-2 1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮
[0070]
白色固体,收率:92%;进行核磁共振检测,检测结果为:1h nmr(300mhz,dmso-d6)δ8.21(s,1h),8.03(d,2h,j=6.0hz),7.98(t,1h,j=9.0hz),7.77(t,1h,j=9.0hz),7.47(d,1h,j=9.0hz),7.34(d,1h,j=9.0hz),4.06(s,3h),2.42(s,3h)。由此可知,所得化合物的结构式为:
实施例6式iii化合物的制备以5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑(iii-1)和5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑(iii-2)为例介绍式iii化合物的合成通法。将化合物1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮或1-(1-甲基-1h-吲唑-5-基)-2-(6-甲基吡啶-2-基)乙烷-1,2-二酮(9.05mmol)溶于甲醇(48ml)中,搅拌下向其中依次加入乙酸铵(89.48mmol),60%的乙二醛二甲基缩醛(2.33ml,13.3mmol)和甲基-叔丁基醚(48ml)后,将反应液加热至50℃反应2h。反应结束后,反应液冷却至室温,用饱和nahco3溶液调ph至中性。反应液用乙酸乙酯萃取3次,合并有机相。合并的有机相用饱和食盐水反复洗涤,加入无水硫酸钠干燥,过滤,减压蒸干。粗品经硅胶柱层析,得固体化合物iii-1和iii-2。化合物iii-15-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑浅黄色固体,收率:64%;进行核磁共振检测,检测结果为:1h nmr(300mhz,dmso-d6)δ13.08(s,1h,nh),12.48(s,1h,nh),8.08(br s,2h),7.60(br s,2h),7.49(d,1h,j=9.0hz),7.06(br s,1h),5.49(s,1h),3.38(s,6h),2.30(s,3h)。由此可知,所得化合物的结构式为:化合物iii-2 5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑浅黄色固体,收率:54%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ10.86(br s,1h,nh),7.99(d,2h,j=9.0hz),7.66(d,1h,j=9.0hz),7.41(d,1h,j=9.0hz),7.35(t,1h,j=7.5hz),7.22(d,1h,j=9.0hz),6.95(d,1h,j=6.0hz),5.58(s,1h),4.10(s,3h),3.45(s,6h),2.53(s,3h)。由此可知,所得化合物的结构式为:实施例7式iv化合物的制备以4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛(iv-1)和4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛(iv-2)为例介绍式iv化合物的合成通法。
将化合物5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1h-吲唑或5-(2-(二甲氧基甲基)-5-(6-甲基吡啶-2-基)-1h-咪唑-4-基)-1-甲基-1h-吲唑(5.74mmol)加入到圆底烧瓶中,再加入1n hcl(18ml),将反应液加热至70℃反应4h。反应结束后,反应液冷却至0℃,用饱和nahco3溶液调ph至中性。生成的固体用布氏漏斗抽滤,用水洗涤滤饼。在减压干燥箱中干燥得化合物iv-1和iv-2。化合物iv-1 4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛浅黄色固体,收率:96%;进行核磁共振检测,检测结果为:1h nmr(300mhz,dmso-d6)δ13.17(s,1h,nh),9.73(s,1h),8.13(s,2h),7.70(t,1h,j=7.5hz),7.61(d,1h,j=9.0hz),7.55(t,2h,j=9.0hz),7.18(d,1h,j=6.0hz),2.37(s,3h)。由此可知,所得化合物的结构式为:化合物iv-2 4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛浅黄色固体,收率:93%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3+dmso-d6)δ9.81(s,1h),8.01(s,2h),7.65(d,1h,j=9.0hz),7.48(d,1h,j=9.0hz),7.41(t,1h,j=7.5hz),7.25(d,1h,j=6.0hz),7.07(d,1h,j=9.0hz),4.31(s,3h),2.58(s,3h)。由此可知,所得化合物的结构式为:实施例8式i化合物的制备以2-取代-4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑(i-1-1-i-1-7)和2-取代-4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑(i-2-1-i-2-7)为例介绍式i化合物的合成通法。将化合物4-(1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛或4-(1-甲基-1h-吲唑-5-基)-5-(6-甲基吡啶-2-基)-1h-咪唑-2-甲醛(0.33mmol)溶于无水四氢呋喃(8ml)中,搅拌下向其中依次加入乙酸(0.02g,0.33mmol)和适当的苯胺类化合物(0.50mmol)后,将反应液加热至75℃反应2h。反应液冷却至0℃,再加甲醇(3ml)。在反应液中分次加入硼氢化钠(0.06g,1.32mmol),然后将反应混合物升至室温,继续搅拌3h后,加水停止反应。将反应液用1n hcl调ph至中性,用ch2cl2萃取3次,合并有机相。合并的有机相用饱和食盐水洗涤,加入无水硫酸钠干燥,过滤,减压蒸干。粗品经硅胶柱层析,得式i化合物。化合物i-1-1 n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺黄色固体,收率:67%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3+dmso-d6)δ7.99(s,1h),7.91(s,1h),7.51(d,2h,j=6.0hz),7.30(t,1h,j=9.0hz),7.18(d,2h,j
=9.0hz),7.12(t,1h,j=7.5hz),6.91(d,1h,j=6.0hz),6.71(d,2h,j=6.0hz),4.47(s,2h),2.13(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
23
h
21
n
6 381.18222,found 381.18253。由此可知,所得化合物的结构式为:化合物i-1-2 n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-氟苯胺白色固体,收率:32%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3+cd3od)δ8.00(s,1h),7.92(s,1h),7.54(d,1h,j=9.0hz),7.48(d,1h,j=9.0hz),7.31(t,1h,j=7.5hz),7.12(d,1h,j=6.0hz),6.99-6.90(m,3h),6.77(t,1h,j=9.0hz),6.68-6.61(m,1h),4.52(s,2h),2.48(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
23
h
20
fn
6 399.17280,found 399.17273。由此可知,所得化合物的结构式为:化合物i-1-3(j-1090)n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-氟苯胺白色固体,收率:40%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3+cd3od)δ7.92(s,1h),7.82(s,1h),7.44(s,2h),7.03(t,2h,j=7.5hz),6.88(d,1h,j=6.0hz),6.43-6.31(m,4h),4.37(s,2h),3.53(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
23
h
20
fn
6 399.17280,found 399.17264。由此可知,所得化合物的结构式为:化合物i-1-4 n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-氟苯胺白色固体,收率:38%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3+cd3od)δ7.92(s,1h),7.82(s,1h),7.44(s,2h),7.27(t,1h,j=9.0hz),7.04(d,1h,j=9.0hz),6.88(d,1h,j=6.0hz),6.79(t,2h,j=7.5hz),6.61-6.57(m,2h),4.35(s,2h),2.43(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
23
h
20
fn
6 399.17280,found 399.17258。由此可知,所得化合物的结构式为:化合物i-1-5 n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-2-甲基苯胺
黄色固体,收率:66%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ8.02(s,1h),7.96(s,1h),7.55(d,1h,j=6.0hz),7.36-7.28(m,2h),7.20(d,1h,j=6.0hz),7.10-7.04(m,2h),6.91(d,1h,j=6.0hz),6.72-6.66(m,2h),4.55(s,2h),2.46(s,3h),2.13(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
23
h
23
n
6 395.19787,found 395.19778。由此可知,所得化合物的结构式为:化合物i-1-6 n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-3-甲基苯胺浅黄色固体,收率:56%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ8.01(s,1h),7.93(s,1h),7.56(d,1h,j=9.0hz),7.49(d,1h,j=9.0hz),7.31(t,1h,j=7.5hz),7.13(d,1h,j=9.0hz),7.06(t,1h,j=7.5hz),6.92(d,1h,j=9.0hz),6.55(t,3h,j=9.0hz),4.48(s,2h),2.50(s,3h),2.25(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
23
h
23
n
6 395.19787,found 395.19806。由此可知,所得化合物的结构式为:化合物i-1-7 n-((4-(1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)-4-甲基苯胺浅黄色固体,收率:60%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ8.04(s,1h),7.96(s,1h),7.58(dd,1h,j=9.0,3.0hz),7.50(d,1h,j=9.0hz),7.33(t,1h,j=7.5hz),7.16(d,1h,j=9.0hz),7.02(d,2h,j=9.0hz),6.94(d,1h,j=9.0hz),6.68(d,2h,j=9.0hz),4.49(s,2h),2.52(s,3h),2.23(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
23
h
23
n
6 395.19787,found 395.19769。由此可知,所得化合物的结构式为:化合物i-2-1 n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺浅黄色固体,收率:60%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ7.97(s,2h),7.65(d,1h,j=9.0hz),7.42(d,1h,j=9.0hz),7.33(t,1h,j=7.5hz),7.19(t,3h,j=7.5hz),6.92(d,1h,j=9.0hz),6.77(t,1h,j=7.5hz),6.70(d,2h,j=9.0hz),4.50(s,2h),4.10(s,3h),2.48(s,3h);hrms-esi(m/z):[m+h]
+ calcd for c
24
h
23
n
6 395.19787,found 395.19794。由此可知,所得化合物的结构式为:
化合物i-2-2 2-氟-n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺浅黄色固体,收率:35%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ7.99(s,1h),7.97(s,1h),7.67(d,1h,j=9.0hz),7.41(d,1h,j=6.0hz),7.34(t,1h,j=7.5hz),7.21(d,1h,j=9.0hz),6.99-6.88(m,3h),6.75(t,1h,j=7.5hz),6.71-6.64(m,1h),4.59(br s,1h,nh),4.51(s,2h),4.10(s,3h),2.40(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
24
h
22
fn
6 413.18845,found 413.18887。由此可知,所得化合物的结构式为:化合物i-2-3 3-氟-n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺浅黄色固体,收率:40%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ7.96(s,2h),7.65(d,1h,j=6.0hz),7.40(d,1h,j=9.0hz),7.35(t,1h,j=9.0hz),7.21(d,1h,j=9.0hz),7.06(dd,1h,j=15.0,6.0hz),6.92(d,1h,j=6.0hz),6.43-6.34(m,2h),6.30-6.25(m,1h),4.60(br s,1h,nh),4.37(s,2h),4.09(s,3h),2.43(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
24
h
22
fn
6 413.18845,found 413.18863。由此可知,所得化合物的结构式为:化合物i-2-4 4-氟-n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺浅黄色固体,收率:38%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ7.98(s,2h),7.65(d,1h,j=9.0hz),7.41(d,1h,j=6.0hz),7.37(t,1h,j=9.0hz),7.22(d,1h,j=6.0hz),6.94(d,1h,j=6.0hz),6.85(t,2h,j=9.0hz),6.58-6.54(m,2h),4.40(s,2h),4.11(s,3h),2.45(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
24
h
22
fn
6 413.18845,found 413.18872。由此可知,所得化合物的结构式为:化合物i-2-5 2-甲基-n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺
浅黄色固体,收率:61%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3+cd3od)δ7.98(s,2h),7.65(d,1h,j=9.0hz),7.40(d,1h,j=6.0hz),7.33(t,1h,j=7.5hz),7.20(d,1h,j=9.0hz),7.12(d,1h,j=9.0hz),7.07(d,1h,j=6.0hz),6.91(d,1h,j=6.0hz),6.72(d,1h,j=6.0hz),6.68(d,1h,j=6.0hz),4.53(s,2h),4.10(s,3h),2.46(s,3h),2.18(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
25
h
25
n
6 409.21352,found 409.21329。由此可知,所得化合物的结构式为:化合物i-2-6 3-甲基-n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺浅黄色固体,收率:58%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ7.96(s,2h),7.65(d,1h,j=9.0hz),7.40(d,1h,j=9.0hz),7.33(t,1h,j=7.5hz),7.20(d,1h,j=9.0hz),7.05(t,1h,j=9.0hz),6.91(d,1h,j=9.0hz),6.58(d,1h,j=9.0hz),6.46(d,2h,j=6.0hz),4.45(s,2h),4.09(s,3h),2.45(s,3h),2.24(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
25
h
25
n
6 409.21352,found 409.21371。由此可知,所得化合物的结构式为:化合物i-2-7 4-甲基-n-((4-(1-甲基-1h-吲唑-5-基)-(6-甲基吡啶-2-基)-1h-咪唑-2-基)甲基)苯胺浅黄色固体,收率:60%;进行核磁共振检测,检测结果为:1h nmr(300mhz,cdcl3)δ7.96(s,2h),7.64(d,1h,j=9.0hz),7.39(d,1h,j=9.0hz),7.33(t,1h,j=7.5hz),7.19(d,1h,j=9.0hz),6.98(d,2h,j=9.0hz),6.91(d,1h,j=9.0hz),6.57(d,2h,j=9.0hz),4.42(s,2h),4.09(s,3h),2.45(s,3h),2.22(s,3h);hrms-esi(m/z):[m+h]
+
calcd for c
25
h
25
n
6 409.21352,found 409.21332。由此可知,所得化合物的结构式为:生物学数据利用下列测定法可以评估本发明化合物的生物学活性:实施例9含吲唑结构的咪唑类衍生物的alk5激酶抑制活性研究alk5激酶磷酸化抑制活性测定方法alk5蛋白来源于sf9昆虫细胞中表达的人类重组gst-融合蛋白。激酶分析使用微孔容
积为50微升的珀金埃尔默的96孔板。将依次加入20μl实验缓冲液(标准缓冲液),5μlatp溶液(在h2o中),5μl测试化合物(溶解于10%dmso中),20μl酶/底物来配制反应混合物。已配制的反应混合物在30℃孵育60min后,放置于50微升2%(v/v)h3po4溶液中,除去测定缓冲液,用200微升0.9%(w/v)氯化钠溶液洗涤2次。采用微平板闪烁计数器建立了
33
pi的掺入。激酶活性分析利用beckman coulter/sagian
t
m core系统来检测。测定结果见表1。表1式i化合物alk5激酶抑制活性表1式i化合物alk5激酶抑制活性式i化合物alk5激酶半数抑制活性(ic
50
)测定结果如表1所示,合成的所有目标化合物的活性远高于阳性对照化合物ly-2157299,其中化合物i-1-1和i-1-3的抑制活性最强。为了证明该类化合物的有效性,选择了alk5抑制活性最好的化合物i-1-3(j-1090)。该方法是对人脑胶质瘤细胞u87按浓度梯度处理j-1090。结果j-1090剂量性抑制tgf-β诱导emt转录因子slug及间充质标志性蛋白(n-cadherin、vimentin),导致下调细胞转移和侵袭能力。许多生长因子在诱导emt方面备受关注,包括肝细胞生长因子hgf、表皮生长因子egf、血小板源性生长因子pdgf、tgf-β等与上皮细胞表面相应受体结合,通过细胞内多种信号通路转导途径将信号转入细胞内,活化不同核内转录因子,调节转导基因的表达,最终促进emt的发生。在下面实例中emt是由tgf-β诱导的。本发明涉及的j-1090的浓度梯度为0.1μm、0.5μm、1.0μm。实施例10细胞培养技术细胞进入对数生长期后,用0.25%胰酶消化,按细胞数5
×
104密度平均分入6个直径为6cm细胞培养皿中,每个培养皿加入3ml dmem培养基(含已灭活的10%小牛血清,100u/l青霉素)放入恒温培养箱(37℃,5%co2,正常氧浓度和饱和湿度)中培养。待细胞完全贴壁后,将细胞分为阴性对照组、阳性对照组、药物浓度梯度组(0.1μm,1μm),将阳性对照组和药物浓度梯度组处理tgf-β3μl(终浓度10ng/ml),培养48h,将药物浓度梯度组(0.1μm,1μm),培养12h。
如图1所示,光学显微镜下观察u87细胞形态证实j-1090以剂量依赖性方式抑制tgf-β诱导的间充质形态,其效果优于阳性对照药ly-2157299。实施例11蛋白质免疫印迹法(western blot法)具体实验步骤如下:(1)蛋白样品的制备,总蛋白的提取。(2)蛋白变性:取样本蛋白15μl分别加入相应的tube中,加入5μl上样缓冲液,煮沸5min。(3)将煮沸后的蛋白样品放置sds-page凝胶上电泳。(4)待电泳停止,将凝胶上的蛋白样品转录至甲醇浸泡的pvdf膜上。(5)将pvdf膜放入5%脱脂牛奶中室温封闭1h。(6)一抗杂交,二抗杂交。(7)ecl发光试剂盒检测。如图2所示,用western blot法证实了j-1090以剂量依赖性方式抑制tgf-β诱导u87细胞核内slug表达,其效果优于阳性对照药ly-2157299。如图3所示,用western blot法证实了j-1090以剂量依赖性方式抑制tgf-β诱导的emt间充质标志物的表达,n-cadherin、vimentin表达下调,其效果优于阳性对照药ly-2157299。实施例12pcr法具体实验步骤如下:(1)总rna提取:将tgf-β和药物处理后的细胞用pbs反复清洗三次,加入1ml ttiol,用注射器反复吹打,使其全部裂解。加入200μl ccl4使溶液分层,4℃12000r离心15min。取上层至新的tube中,加入500μl异丙醇,混匀,4℃12000r离心10min。小心弃去上清液,在含rna的tube中加入1ml 75%乙醇,振荡,4℃12000r离心5min。充分弃去上清液,放置超净台通风干燥。待彻底干燥后,加入30μl depc-ddw,使之完全溶解。(2)以rna为模板逆转录合成cdna:取出提取的mrna样品,标记新的pcr离心管,加入以下试剂:3μl mrna、1μl anchored-oligo(dt)
18
引物、9μl三蒸水,将以上试剂混匀,用离心机轻微离心,放到pcr仪上,温度设为65℃,时间设定为10min,加热结束后,取出离心管,置于冰上冷却,然后继续加入以下试剂:4μl 5
×
rt buffer、0.5μl rnase inhibitor(20u/μl)、2μl dntp mixture、0.5μl逆转录酶。并设置反应条件为55℃30min、85℃5min。反应结束后,将产物放在冰上冷却,用于接下来pcr实验。(3)pcr反应:由cdna合成双链dna,取pcr离心管,加入以下试剂:1μl引物(正)、1μl引物(反)、2μl dntp、2.5μl 10
×
pcr buffer、0.2μl taq dna ploymerase、3μl cdna、15.3μl三蒸水,将以上试剂混匀,用离心机轻微离心,放到pcr仪上,pcr条件为94℃4min(预变性)、94℃30s、58℃30s、72℃30s、72℃5min为30~40循环,注意循环中不包含预变性,具体视反应引物而定。如图4所示,用pcr法证实了j-1090抑制tgf-β诱导的emt间充质标志物mrna水平。实施例13
免疫荧光法具体实验步骤如下:(1)取生长对数期的细胞,接种至铺盖玻片的24孔板中。(2)将tgf-β和药物处理后的细胞在盖玻片上生长融合95%~100%时,从培养箱中取出,用预温pbs洗涤3次,10min/次。(3)4%的甲醛室温固定30min。pbs洗涤3次,10min/次。(4)0.2%triton x-100透化5min。pbs洗涤3次,10min/次。(5)5%bsa室温封闭30min。加一抗slug/n-cadherin(用1%bsa稀释)孵育,4℃过夜。pbs洗涤3次,10min/次。(6)加二抗mouse-488(用1%bsa稀释)孵育30min,避光。pbs洗涤3次,10min/次。(7)染dapi(终浓度1μg/ml)30min,避光。(8)95%甘油封片。如图5所示,用免疫荧光法证实了j-1090几乎完全阻断tgf-β诱导u87细胞核内slug表达。图中
“-”
代表阴性对照组,“+”代表阳性对照组。如图6所示,用免疫荧光法证实了j-1090几乎完全阻断tgf-β诱导u87细胞n-cadherin表达。图中
“-”
代表阴性对照组,“+”代表阳性对照组。实施例14细胞划痕实验具体实验步骤如下:(1)取对数生长期u87细胞接种于6孔板,待长到80%以上,换成free dmem饥饿细胞12h,tgf-β诱导组,再用含tgf-β(10ng/ml)无血清培养基诱导48h。(2)用10μl枪头尖在6孔板上平行划痕,用pbs冲掉刮下的细胞,并用显微镜拍摄划痕的宽度(0h)。(3)换dmem培养基,药物处理组加j-1090(1μm/ml)培养15h、36h,测量划痕宽度并拍照。如图7所示,划痕法证实了j-1090抑制tgf-β增加的肿瘤细胞转移能力。实施例15细胞入侵实验具体实验步骤如下:(1)transwell小室(聚碳酸酯膜微孔径8μm)内加入稀释的matrigel 100μl。将transwell小室插入24孔板,37℃恒温培养箱孵育24h。(2)取对数生长期u87细胞浓度调整为2.5
×
105ml细胞悬浮在200ml无血清培养基,加入transwell小室的上室面,小室的下室内加入700ml含10%fbs培养基作为趋化剂。(3)置于恒温细胞培养箱培养24h后,取出transwell小室,吸弃培养基,用棉签擦去上室面未穿过膜的细胞,pbs清洗3次。(4)4%多聚甲醛固定细胞30min后,0.1%结晶紫染色5min,pbs清洗2次,在显微镜下每个样本随机选取5个视野,拍照记录实验结果。如图8所示,入侵法证实了j-1090抑制tgf-β增加的肿瘤细胞侵袭能力。以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为
本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips