
 商标分类
商标分类  商标转让
商标转让 
一种3-氨基-4-氯苯甲酸十六酯的制备方法与流程
 2021-02-02 07:02:11|
2021-02-02 07:02:11| 385|
385| 起点商标网
起点商标网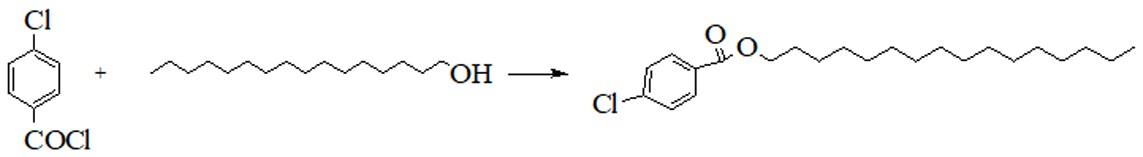
[0001]
本发明涉及一种化合物的制备方法,尤其涉及一种3-氨基-4-氯苯甲酸十六酯的制备方法,属于化合物制备技术领域。
背景技术:
[0002]
3-氨基-4-氯苯甲酸十六酯是一种重要的感光材料的中间体,以该产品为原料合成的感光材料具有较高的活性和照相性能,为摄影爱好者提供了更多更好的选择,同时也为摄影艺术的创作提供了广阔的空间,因其需求量大,市场前景看好。
[0003]
3-氨基-4-氯苯甲酸十六酯的合成方法主要有以下4种:(1)以3-氨基-4-氯苯甲酸为原料,在四氯化锡或氯化亚锡催化作用下与十六醇进行酯化反应(2)以3-氨基-4-氯苯甲酸为原料,在碱性条件下与溴代十六烷进行酯化反应(3)以3-硝基-4-氯苯甲酸为原料,先与十六醇反应,再与氢气在钯碳催化下进行还原反应(4)以十六醇为原料,先与磺酰氯反应,再与3-氨基-4-氯苯甲酸进行酯交换反应。
[0004]
上述几种合成方法都存在缺陷,方法(1)和(3)需要十六醇大大过量,同时产品需要重结晶进行精制,生产比较繁琐;方法(2)所用溴代十六烷价格昂贵,导致产品成本高,在市场上不具备竞争优势;方法(4)副反应较多,同时反应不完全,导致产品纯度低、收率低,也不具备优势。
技术实现要素:
[0005]
为了解决上述技术问题,本发明的目的在于提供一种制备成本低且收率高、产品纯度高的3-氨基-4-氯苯甲酸十六酯的制备方法。
[0006]
为了实现上述技术目的,本发明提供了一种3-氨基-4-氯苯甲酸十六酯的制备方法,该制备方法包括以下步骤:将氯化剂、对氯苯甲酸和n,n-二甲基甲酰胺混合,进行酰氯化反应,得到对氯苯甲酰氯(如图1所示);向对氯苯甲酰氯中加入十六醇和第一有机溶剂,进行酯化反应,当十六醇含量<0.5%时,停止反应,降温析晶,经抽滤、干燥,得到对氯苯甲酸十六酯(如图2所示);向对氯苯甲酸十六酯中加入第二有机溶剂,搅拌的同时滴加硫酸与硝酸,通过调节滴加速度控制温度在50℃以下;滴加结束,进行硝化反应;经降温、抽滤、水洗,得到3-硝基-4-氯苯甲酸十六酯(如图3所示);向3-硝基-4-氯苯甲酸十六酯中加入第三有机溶剂和催化剂,氮气置换,氢气置换,然后充氢气进行还原反应;反应结束后静置,将上层液体通过氮气压出高压釜,减压回收溶剂,降温析晶,抽滤、烘干,得到3-氨基-4-氯苯甲酸十六酯(如图4所示)。
[0007]
本发明的3-氨基-4-氯苯甲酸十六酯的制备方法,以对氯苯甲酸为起始原料,先与dmf、氯化试剂进行酰氯化反应,生成对氯苯甲酰氯;再与十六醇进行酯化反应,生成对氯苯甲酸十六酯;再与硫酸、硝酸进行硝化反应,生成3-硝基-4-氯苯甲酸十六酯;最后与氢气在
雷尼镍催化下进行还原反应,生成3-氨基-4-氯苯甲酸十六酯。四步总收率高达86%,产品纯度高达99.5%。
[0008]
在本发明的一具体实施方式中,进行酰氯化反应时,氯化剂、对氯苯甲酸和n,n-二甲基甲酰胺的摩尔比为1.0-3.5:1.0:0-0.15。
[0009]
在本发明的一具体实施方式中,进行酰氯化反应时,氯化剂为三氯化磷、三氯氧磷或氯化亚砜。
[0010]
在本发明的一具体实施方式中,进行酰氯化反应时,酰氯化反应的反应温度为40℃-90℃(优选为50℃-80℃),酰氯化反应的反应时间为2小时-10小时(优选为4小时-8小时)。
[0011]
在本发明的一具体实施方式中,进行酯化反应时,对氯苯甲酰氯、十六醇和第一有机溶剂的摩尔比为1.0:1.0-1.2:1.5-3.5。
[0012]
在本发明的一具体实施方式中,进行酯化反应时,第一有机溶剂为二氯乙烷、四氯乙烯或1,3-二氯丙烷。
[0013]
在本发明的一具体实施方式中,进行酯化反应时,酯化反应的反应温度为30℃-100℃(优选为50℃-90℃),反应时间为1小时-8小时(优选为2小时-6小时),抽滤温度为-5℃至30℃(优选为0℃-10℃)。
[0014]
在本发明的一具体实施方式中,进行硝化反应时,滴加硫酸与硝酸的温度为20℃-50℃(优选为20℃-30℃)。
[0015]
在本发明的一具体实施方式中,进行硝化反应时,对氯苯甲酸十六酯中、第二有机溶剂的摩尔比为1.0:7.0-14.0;硫酸、硝酸与对氯苯甲酸十六酯的摩尔比为0.1-1.0:1.0-2.0:1(优选为0.4-0.8:1.2-1.6:1)。
[0016]
在本发明的一具体实施方式中,进行硝化反应时,第二有机溶剂为二氯乙烷、四氯乙烯或1,3-二氯丙烷。
[0017]
在本发明的一具体实施方式中,进行硝化反应时,硝化反应的反应温度为60℃-90℃,反应时间为2小时-5小时,抽滤温度为10℃-25℃。
[0018]
在本发明的一具体实施方式中,进行硝化反应时,根据温度要求来调节滴加的速度,该反应放热明显,只要不超过50℃就可以加快滴加速度,反之亦然。
[0019]
在本发明的一具体实施方式中,进行还原时,3-硝基-4-氯苯甲酸十六酯、第三有机溶剂和催化剂的重量比为1.0:2.0-2.5:0.01-0.1。
[0020]
在本发明的一具体实施方式中,进行还原时,第三有机溶剂为甲醇、乙醇或异丙醇。
[0021]
在本发明的一具体实施方式中,进行还原时,反应的温度为50℃-80℃,保压压力为0.1mpa-1.5mpa(0.5mpa-1.5mpa),反应时间为1-8小时。
[0022]
在本发明的一具体实施方式中,进行还原时,采用的催化剂为雷尼镍催化剂;优选地,雷尼镍催化剂的重量为3-硝基-4-氯苯甲酸十六酯重量的0.01倍-0.10倍(优选为0.01倍-0.03倍)。
[0023]
在本发明的一具体实施方式中,反应结束后,减压回收溶剂时,温度<60℃,真空在-0.09mpa以上。
[0024]
本发明的3-氨基-4-氯苯甲酸十六酯的制备方法,以对氯苯甲酸为起始原料,先与
dmf、氯化试剂进行酰氯化反应,生成对氯苯甲酰氯;再与十六醇进行酯化反应,生成对氯苯甲酸十六酯;再与硫酸、硝酸进行硝化反应,生成3-硝基-4-氯苯甲酸十六酯;最后与氢气在雷尼镍催化下进行还原反应,生成3-氨基-4-氯苯甲酸十六酯。具体包括以下步骤:1)在反应器中加入氯化剂、对氯苯甲酸和n,n-二甲基甲酰胺,然后升温进行保温反应,反应结束后先真空脱溶,回收多余的氯化剂待下次套用,再用高真空脱去残留的微量氯化剂,残余物即为对氯苯甲酰氯;2)向步骤1)得到的对氯苯甲酰氯中加入十六醇和第一有机溶剂,升温进行酯化反应,在保温反应过程中取样检测,当十六醇含量(gc)<0.5%时,停止反应,降温析晶,经抽滤、干燥,得到对氯苯甲酸十六酯;3)向步骤2)得到的对氯苯甲酸十六酯中加入第二有机溶剂,开启搅拌,滴加硫酸与硝酸,调节滴加速度,控制温度在50℃以下;滴加结束,保温搅拌反应;反应结束后加水回收第二有机溶剂,回收完毕,经降温、抽滤、水洗,得到3-硝基-4-氯苯甲酸十六酯;4)向步骤3)得到的3-硝基-4-氯苯甲酸十六酯中加入第三有机溶剂和雷尼镍催化剂,先氮气置换3次,再用氢气置换3次,然后充氢气保温保压反应;反应结束后静置,将上层液体通过氮气压出高压釜,减压回收溶剂,然后降温析晶,抽滤、烘干,得到3-氨基-4-氯苯甲酸十六酯。
[0025]
本发明的3-氨基-4-氯苯甲酸十六酯的制备方法,避免了十六醇的大大过量,省去了回收十六醇的步骤,简化了生产操作;同时采用对氯苯甲酰氯与十六醇进行酯化反应,反应更彻底,基本无副反应,纯度更高,产品无需精制,避免了以往合成方法产品需要精制提纯的麻烦,大大节约了成本。
[0026]
本发明的3-氨基-4-氯苯甲酸十六酯的制备方法的总收率达86%,产品纯度达99.5%。以对氯苯甲酸为起始原料,原料易得且价格低廉;十六醇用量大幅降低,控制在1.02eq,省去了回收十六醇的麻烦;设计更加合理的合成路线,副反应少,杂质小,产品无需重结晶精制,一次性合格;产品纯度更高,成本更低。
附图说明
[0027]
图1为本发明制备对氯苯甲酰氯的合成路线示意图。
[0028]
图2为本发明制备对氯苯甲酸十六酯的合成路线示意图。
[0029]
图3为本发明制备3-硝基-4-氯苯甲酸十六酯的合成路线示意图。
[0030]
图4为本发明制备3-氨基-4-氯苯甲酸十六酯的合成路线示意图。
[0031]
图5为实施例1中的对氯苯甲酰氯的核磁氢谱-1
h nmr。
[0032]
图6为实施例1中的对氯苯甲酸十六酯的核磁氢谱-1
h nmr。
[0033]
图7为实施例1中的3-硝基-4-氯苯甲酸十六酯的核磁氢谱-1
h nmr。
[0034]
图8为实施例1中的3-氨基-4-氯苯甲酸十六酯的核磁氢谱-1
h nmr。
具体实施方式
[0035]
实施例中gc中控分析方法:色谱柱:非极性色谱柱(db-1ms、30m
×
0.25mm
×
0.25um)或等类型柱子;进样器温度:340℃;
检测器温度:340℃;柱子温度:70℃(保持3min)
→
20℃/min
→
300℃(保持25min);流速=1.0ml/min;溶剂:二氯甲烷;浓度:约20mg/ml;进样量:1.0ul。
[0036]
实施例1在四口瓶中投入氯化亚砜:178.5g(1.5mol)、对氯苯甲酸:78.2g(0.5mol),dmf:4g(0.055mol),打开搅拌,缓慢升温至60-65℃,在此温度下保温反应4小时,反应结束,水泵真空脱溶,回收未反应的氯化亚砜,套用至下批反应;然后切换至油泵继续脱除残留的少量氯化亚砜,残余物即为对氯苯甲酰氯:约90g,即中间产物ⅰ,纯度99.2%。对氯苯甲酰氯的核磁氢谱-1
h nmr如图5所示,1h nmr(cdcl3),δ=7.48(d,2h,arh),8.01(d,2h,arh)。
[0037]
在四口瓶中加入对氯苯甲酰氯:90g和二氯乙烷:150g,再加入十六醇:123.4g(0.51mol)和二氯乙烷:150g,缓慢升温至60-65℃,并在此温度下保温反应6小时,取样检测,十六醇剩余0.3%(gc),停止反应,降温析晶,降至5-10℃抽滤,烘干,得到对氯苯甲酸十六酯:183.1g,即中间产物ⅱ,纯度99.6%。对氯苯甲酸十六酯的核磁氢谱-1
h nmr如图6所示,1h nmr(cdcl3),δ=0.96(t,3h,ch3),1.29(m,24h,ch2),1.33(m,2h,ch2),1.66(m,2h,ch2),3.96(t,2h,ch2),7.38(d,2h,arh),7.91(d,2h,arh)。
[0038]
在四口瓶中加入对氯苯甲酸十六酯:183.1g,二氯乙烷:600g,调整温度在20-30℃,开始滴加配制好的混酸:浓硫酸:20g(0.204mol)+发烟硝酸:50g(0.754mol),滴加期间控制温度低于50℃,滴加结束升温至60-65℃并在此温度下保温反应5小时,反应完毕,降温至40℃以下缓慢加入水:800g,然后开始真空脱溶,回收二氯乙烷,回收完毕,降温至20-25℃,抽滤,水洗,得到3-硝基-4-氯苯甲酸十六酯湿品:约240g,即中间产物ⅲ,纯度99.3%。3-硝基-4-氯苯甲酸十六酯的核磁氢谱-1
h nmr如图7所示,1h nmr(cdcl3),δ=0.96(t,3h,ch3),1.29(m,24h,ch2),1.33(m,2h,ch2),1.75(m,2h,ch2),4.25(t,2h,ch2),7.64(d,1h,arh),8.30(d,1h,arh),8.84(s,1h,arh)。
[0039]
在高压釜里中加入3-硝基-4-氯苯甲酸十六酯湿品:240g,甲醇:500g,雷尼镍催化剂:5.0g,加完物料,合上釜盖,检测气密性;确认高压釜气密性良好的情况下,先用氮气置换3次,再用氢气置换3次,最后通氢气维持釜内压力在1.2-1.5mpa,开始升温,控制釜内温度在55-65℃,当釜内压力不再下降时,继续保温反应1小时,停止搅拌,静置1小时,将上层清夜用氮气压出高压釜,加入活性炭:4g在50℃左右保温脱色0.5小时,过滤,滤液真空脱溶至有固体析出,然后降温至0-5℃抽滤,烘干,得到3-氨基-4-氯苯甲酸十六酯:171.8g,纯度99.56%。3-氨基-4-氯苯甲酸十六酯的核磁氢谱-1
h nmr如图8所示,1h nmr(cdcl3),δ=0.96(t,3h,ch3),1.29(m,24h,ch2),1.33(m,2h,ch2),1.75(m,2h,ch2),4.0(s,2h,nh2),4.25(t,2h,ch2),7.11(s,1h,arh),7.13(d,1h,arh),7.27(d,1h,arh)。
[0040]
实施例2在四口瓶中投入三氯氧磷:153.5g(1.0mol)、对氯苯甲酸:78.2g(0.5mol),dmf:3g(0.041mol),打开搅拌,缓慢升温至50~55℃,在此温度下保温反应5小时,反应结束,水泵真空脱溶,回收未反应的三氯氧磷,套用至下批反应;然后切换至油泵继续脱除残留的少量三
氯氧磷,残余物即为对氯苯甲酰氯:约92g,即中间产物ⅰ,纯度99.3%。
[0041]
在四口瓶中加入对氯苯甲酰氯:92g和四氯乙烯:150g,再加入十六醇:123.4g(0.51mol)和四氯乙烯:150g,缓慢升温至80~85℃,并在此温度下保温反应3小时,取样检测,十六醇剩余0.26%(gc),停止反应,降温析晶,降至10~15℃抽滤,烘干,得到对氯苯甲酸十六酯:183.4g,即中间产物ⅱ,纯度99.5%。
[0042]
在四口瓶中加入对氯苯甲酸十六酯:183.4g,四氯乙烯:600g,调整温度在20-30℃,开始滴加配制好的混酸:浓硫酸:30g(0.306mol)+发烟硝酸:40g(0.603mol),滴加期间控制温度低于50℃,滴加结束升温至75-80℃并在此温度下保温反应3小时,反应完毕,降温至40℃以下缓慢加入水:800g,然后开始真空脱溶,回收四氯乙烯,回收完毕,降温至20-25℃,抽滤,水洗,得到3-硝基-4-氯苯甲酸十六酯湿品:约239g,即中间产物ⅲ,纯度99.2%。
[0043]
在高压釜里中加入3-硝基-4-氯苯甲酸十六酯湿品:239g,异丙醇:500g,雷尼镍催化剂:3.0g,加完物料,合上釜盖,检测气密性;确认高压釜气密性良好的情况下,先用氮气置换3次,再用氢气置换3次,最后通氢气维持釜内压力在0.6-0.9mpa,开始升温,控制釜内温度在70-80℃,当釜内压力不再下降时,继续保温反应1小时,停止搅拌,静置1小时,将上层清夜用氮气压出高压釜,加入活性炭:4g在50℃左右保温脱色0.5小时,过滤,滤液真空脱溶至有固体析出,然后降温至0-5℃抽滤,烘干,得到3-氨基-4-氯苯甲酸十六酯:172.0g,纯度99.65%。
[0044]
实施例3在四口瓶中投入三氯化磷:165.0g(1.2mol)、对氯苯甲酸:78.2g(0.5mol),dmf:2g(0.027mol),打开搅拌,缓慢升温至75-80℃,在此温度下保温反应3小时,反应结束,水泵真空脱溶,回收未反应的三氯化磷,套用至下批反应;然后切换至油泵继续脱除残留的少量三氯化磷,残余物即为对氯苯甲酰氯:约91g,即中间产物ⅰ,纯度99.4%。
[0045]
在四口瓶中加入对氯苯甲酰氯:91g和1,3-二氯丙烷:150g,再加入十六醇:123.4g(0.51mol)和1,3-二氯丙烷:150g,缓慢升温至70-75℃,并在此温度下保温反应4小时,取样检测,十六醇剩余0.2%(gc),停止反应,降温析晶,降至0-5℃抽滤,烘干,得到对氯苯甲酸十六酯:183.3g,即中间产物ⅱ,纯度99.7%。
[0046]
在四口瓶中加入对氯苯甲酸十六酯:183.3g,1,3-二氯丙烷:600g,调整温度在20-30℃,开始滴加配制好的混酸:浓硫酸:35g(0.357mol)+发烟硝酸:45g(0.678mol),滴加期间控制温度低于50℃,滴加结束升温至65-70℃并在此温度下保温反应4小时,反应完毕,降温至40℃以下缓慢加入水:800g,然后开始真空脱溶,回收二氯乙烷,回收完毕,降温至20-25℃,抽滤,水洗,得到3-硝基-4-氯苯甲酸十六酯湿品:约238g,即中间产物ⅲ,纯度99.5%。
[0047]
在高压釜里中加入3-硝基-4-氯苯甲酸十六酯湿品:238g,乙醇:500g,雷尼镍催化剂:4.0g,加完物料,合上釜盖,检测气密性;确认高压釜气密性良好的情况下,先用氮气置换3次,再用氢气置换3次,最后通氢气维持釜内压力在0.8-1.2mpa,开始升温,控制釜内温度在50-55℃,当釜内压力不再下降时,继续保温反应1小时,停止搅拌,静置1小时,将上层清夜用氮气压出高压釜,加入活性炭:4g在50℃左右保温脱色0.5小时,过滤,滤液真空脱溶至有固体析出,然后降温至0-5℃抽滤,烘干,得到3-氨基-4-氯苯甲酸十六酯:171.5g,纯度99.61%。
[0048]
对比例1
本对比例的制备方法基本与实施例1相同,区别在于:酰氯化反应的反应温度为100℃,反应时间为1小时;得到对氯苯甲酰氯:约86g,纯度97.5%。
[0049]
对比例2本对比例的制备方法基本与实施例1相同,区别在于:采用对比例1的对氯苯甲酰氯;酯化反应的反应温度为150℃,反应时间为10小时,抽滤温度为40℃。得到对氯苯甲酸十六酯:约180g,纯度98.5%。
[0050]
对比例3本对比例的制备方法基本与实施例1相同,区别在于:采用对比例2的对氯苯甲酸十六酯;硝化反应时,滴加硫酸与硝酸的温度为60℃;硝化反应的温度为100℃,反应时间为8小时,抽滤温度为30℃。得到3-硝基-4-氯苯甲酸十六酯约235g,纯度97.8%。
[0051]
对比例4本对比例的制备方法基本与实施例1相同,区别在于:采用对比例3的3-硝基-4-氯苯甲酸十六酯;还原反应的温度为90℃,保压压力为3mpa。得到3-氨基-4-氯苯甲酸十六酯:约168g,纯度98.5%。
[0052]
本发明的制备方法的总收率高达86%,产品纯度达99.5%。本发明以对氯苯甲酸为起始原料,原料易得且价格低廉,很适合工业化生产;解决了以往十六醇大大过量需要回收套用的问题,操作更简单;设计更加合理的合成路线,减少了副反应,杂质小,产品无需重结晶精制,纯度一次性达到99.5%。
[0053]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips