
 商标分类
商标分类  商标转让
商标转让 
改造的CASCADE组分和CASCADE复合体的制作方法
 2021-02-02 06:02:28|
2021-02-02 06:02:28| 314|
314| 起点商标网
起点商标网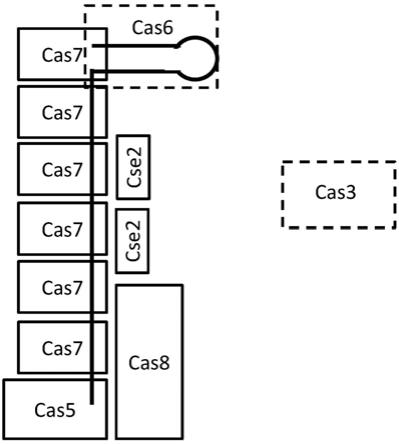
改造的cascade组分和cascade复合体
[0001]
相关申请的交叉引用
[0002]
本申请是2019年5月22日递交的现在待决的第16/420,061号美国专利申请系列的部分延续,其中第16/420,061号美国专利申请系列是2019年1月30日递交的现在授权的第16/262,773号美国专利申请系列的延续,第16/262,773号美国专利申请系列是2018年8月17日递交的现在是2019年3月12日发布的第10,227,576号美国专利的第16/104,875号美国专利申请系列的延续,并且本申请要求2018年6月13日递交的现在待决的第62/684,735号美国临时专利申请系列以及2019年2月19日递交的现在待决的第62/807,717号美国临时专利申请系列的权益:以上申请系列的内容通过引用整体并入本文。
[0003]
关于联邦赞助的研究或开发的申明
[0004]
不适用。
[0005]
序列表
[0006]
本申请含有以ascii形式电子提交的序列表,并在此通过引用整体并入本文中。于2019年6月12日生成的ascii拷贝命名为cbi032-30_st25.txt,且大小为3.1mb。
技术领域
[0007]
本公开总体上涉及改造的第一类i型crispr-cas(cascade)系统,其包括多蛋白效应子复合体、包含i型crispr-cas亚基蛋白和核酸向导的核蛋白复合体、编码i型crispr-cas亚基蛋白的多核苷酸以及向导多核苷酸。本公开还涉及用于制备和使用本发明的改造的i型crispr-cas系统的组合物和方法。
背景技术:
[0008]
规律间隔成簇短回文重复序列(crispr)和crispr相关蛋白(cas)构成了crispr-cas系统。crispr-cas系统提供了针对细菌和古菌中的外来多核苷酸的适应性免疫(参见,例如barrangou,r.,et al.,science 315:1709-1712(2007);makarova,k.s.,et al.,nature reviews microbiology 9:467-477(2011);garneau,j.e.,et al.,nature 468:67-71(2010);sapranauskas,r.,et al.,nucleic acids res.39:9275-9282(2011);koonin,e.v.,et al.,curr.opin.microbiol.37:67-78(2017))。各种crispr-cas系统在其天然宿主中能够靶向dna(第一类i型;第二类ii型和v型)、靶向rna(第二类vi型),以及联合靶向dna和rna(第一类iii型)(参见,例如makarova,k.s.,et al.,nat.rev.microbiol.13:722-736(2015);shmakov,s.,et al.,nat.rev.microbiol.15:169-182(2017);abudayyeh,o.o.,et al.,science 353:1-17(2016))。
[0009]
crispr-cas系统的分类已拥有许多细分级数。koonin,e.v.,et al.,(curr.opin.microbiol.37:67-78(2017))提出了一种分类系统,该系统考虑了对crispr-cas系统的各个类型和亚型具有特异性的标签cas基因。该分类还考虑了多个共有cas蛋白之间的序列相似性、最佳保守cas蛋白的系统发生、基因组织以及crispr阵列的结构。该方法提供了一种分类方案,其将crispr-cas系统分成两种不同的类型:第一类包括多蛋白效
应子复合体(i型(用于抗病毒防御的crispr相关复合体(“cascade”)效应子复合体)、iii型(cmr/csm效应子复合体)和iv型);并且第二类包括单个效应子蛋白(ii型(cas9)、v型(cas12a,先前称为cpf1)和vi型(cas13a,先前称为c2c2))。在第一类系统中,i型是最常见和多样化的,iii型在古菌中比细菌中更常见,并且iv型是最少见的。
[0010]
i型系统包括标签cas3蛋白。cas3蛋白具有负责dna靶序列切割的解旋酶和dna酶域。迄今为止,已鉴定了i型系统的7种亚型(即i-a、i-b、i-c、i-d、i-e、i-f型(以及i-f(例如,i-fv1、i-fv2))和i-u的变体),其具有可变数量的cas基因。i型cas基因包括但不限于如下基因:cas7、cas5、cas8、cse2、csa5、cas3、cas2、cas4、cas1和cas6。具有i型系统的生物体的实例如下:i-a,闪烁古生球菌(archaeoglobus fulgidus);i-b,克氏梭菌(clostridium kluyveri);i-c,嗜碱耐盐芽孢杆菌(bacillus halodurans);i-u,硫还原地杆菌(geobacter sulfurreducens);i-d,蓝丝菌(cyanothece sp.)8802;i-e,大肠杆菌k12(e.coli k12);i-f,假结核耶尔森氏菌(yersinia pseudo-tuberculosis);i-f变体,腐败希瓦氏菌(shewanella putrefaciens)cn-32(koonin,e.v.,et al.,curr.opin.microbiol.37:67-78(2017))。已描述了cas3蛋白介导的dna切割和渐进性降解的特征(参见,例如plagens,a.,et al.,nucleic acids res.42:5125-5138(2014);maier,l.,et al.,rnabiol.10:865
–
874(2013);hochstrasser,m.,et al.,proc.natl.acad.sci.usa 111:6618
–
6623(2014);sinkunas,t.,et al.,embo j.30:1335
–
1342(2011);westra,e.,et al.,mol.cell 46:595
–
605(2012);mulepati,s.,et al.,j.biol.chem.288:22184
–
22192(2013);sinkunas,t.,et al.,embo j.32:385
–
394(2013);mulepati,s.,et al.,j.biol.chem.288:22184
–
22192(2013);redding,s.,et al.,cell 163:854
–
865(2015);sinkunas,t.,et al.,embo j.32:385
–
394(2013);westra,e.,et al.,mol.cell 46:595
–
605(2012))。
[0011]
i型系统通常编码与crispr rna(crrna或“向导rna”)组合形成cascade复合体的蛋白。这些复合体包含多种蛋白和crrna,这些蛋白和crrna均从该crispr位点转录。在i型系统中,crrna前体的初步加工由cas6催化。这通常导致产生crrna,其具有8个核苷酸的5
′
柄、间隔区和3
′
柄;5
’
柄和3
’
柄均来源于重复的序列。在一些系统中,3
′
柄形成茎-环结构;在其他系统中,crrna的3
′
端的二次加工由核糖核酸酶催化(参见,例如van der oost,j.,et al.,nature reviews microbiology 12:479-492(2014))。
[0012]
i型crispr-cas系统的cascade效应子复合体包含具有旁系同源的重复序列相关未知蛋白(ramps;例如cas7和cas5蛋白)的骨架,所述蛋白含有rna识别基序(rrm)折叠以及另外的“大”和“小”亚基蛋白(参见,例如koonin,e.v.,et al.,curr.opin.microbiol.37:67-78,(2017),图2)。这些cascade效应子复合体通常具有cas5亚基蛋白和数种cas7亚基蛋白。此类cascade效应子复合体还包含向导rna。cascade效应子复合体包含以非对称形式沿向导rna的长度布置的不同的亚基蛋白。cas5亚基蛋白和大亚基蛋白(cas8蛋白)位于复合体的一端处,包裹住向导rna的5
’
端。小亚基蛋白的数个拷贝与结合至cas7亚基蛋白的多个拷贝的向导rna骨架接触。cas6亚基蛋白(另一种ramp蛋白)主要通过与crrna的3
′
柄(重复区)的结合而与cascade效应子复合体结合。cas6亚基蛋白通常用作参加crrna前体加工的重复序列特异性的rna酶;然而,在i-c型系统中,cas5用作重复序列特异性的rna酶,并且不存在cas6。
[0013]
crispr-cas i型cascade亚基蛋白的主要序列具有很小的序列同一性;然而,同源ramp模块的存在和多蛋白效应子复合体的整体结构相似性支持了这些效应子复合体的共同起源(参见,例如koonin,e.v.,et al.,curr.opin.microbiol.37:67-78(2017))。
[0014]
i型crispr-cas系统中的适应性免疫作用机制主要涉及三个阶段:适应、表达和干预。在适应阶段,外来dna或rna感染宿主,并且由各种cas基因编码的蛋白结合感染dna或rna的区域。这样的区域称为前间隔区。前间区序列邻近基序(pam)是与前间隔区相邻的短核苷酸序列(例如2-6个碱基对的dna序列)。pam序列通常由cas1亚基蛋白/cas2亚基蛋白复合体识别,其中活性pam感应位点与cas1亚基蛋白相关(参见,例如jackson,s.a.,et al.,science 356:356(6333)(2017))。
[0015]
在表达阶段,将包含多个间隔区重复元件的crispr阵列转录为单个转录物。单个间隔区重复元件被核酸内切酶(例如i型,cas6蛋白;和i-c型,cas5蛋白)加工成单个crrnas。cas亚基蛋白被表达并与crrna结合形成cascade效应子复合体。
[0016]
cascad效应子复合体扫描感染宿主的外来多核苷酸,以识别与间隔区互补的dna。在i型系统中,当效应子复合体识别出与临近pam的间隔区互补的序列时,就会发生干扰;并且将cas3蛋白募集到与dna结合的cascade效应子复合体,以切割并渐进性地消化外来多核苷酸。
[0017]
makarova,k.s.,et al.,(cell 168:946(2017))提供了i型crispr-cas系统的基因、同系物、cascade复合体以及作用机制的概要信息。
[0018]
因此,i型crispr-cas系统目前在真核基因组改造应用中的用途有限,部分是由于cascade复合体难于异源表达,以及i型crispr-cas系统切割dna靶标的方式。
[0019]
发明概述
[0020]
本发明总体上涉及组合物,其包含改造的i型crispr-cas效应子复合体及其组分包括蛋白质组分、修饰的或差异化改变的向导多核苷酸,以及以上的组合。
[0021]
本发明的一个实施方案是组合物,其包含:
[0022]
第一改造的i型crispr-cas效应子复合体,其包含:
[0023]
第一cse2亚基蛋白、第一cas5亚基蛋白、第一cas6亚基蛋白和第一cas7亚基蛋白,
[0024]
包含第一cas8亚基蛋白和第一foki的第一融合蛋白,其中第一cas8亚基蛋白的n端或第一cas8亚基蛋白的c端通过第一连接子多肽分别与第一foki的c端或n端共价连接,并且其中第一连接子多肽具有10个氨基酸至40个氨基酸的长度,和
[0025]
包含能够结合第一核酸靶序列的第一间隔区的第一向导多核苷酸;以及
[0026]
第二改造的i型crispr-cas效应子复合体,其包含:
[0027]
第二cse2亚基蛋白、第二cas5亚基蛋白、第二cas6亚基蛋白和第二cas7亚基蛋白,
[0028]
包含第二cas8亚基蛋白和第二foki的第二融合蛋白,其中第二cas8亚基蛋白的n端或第二cas8蛋白的c端通过第二连接子多肽分别与第二foki的c端或n端共价连接,并且其中第二连接子多肽具有10个氨基酸至40个氨基酸的长度,和
[0029]
包含能够结合第二核酸靶序列的第二间隔区的第二向导多核苷酸,其中第二核酸靶序列的前间区序列邻近基序(pam)和第一核酸靶序列的pam具有20个碱基对至42个碱基对的间隔区间距。
[0030]
在一些实施方案中,第一连接子多肽和/或第二连接子多肽的长度是15个氨基酸
至30个氨基酸或者17个氨基酸至20个氨基酸的长度。在一个实施方案中,第一连接子多肽和第二连接子多肽的长度是相同的。
[0031]
第二核酸靶序列和第一核酸靶序列之间的间隔区间距包括但不限于22个碱基对至40个碱基对、26个碱基对至36个碱基对、29个碱基对至35个碱基对,或者30个碱基对至34个碱基对。
[0032]
第一foki和第二foki可以是能够结合形成同型二聚体的单体亚基,或者是能够结合形成异型二聚体的不同的亚基。
[0033]
在一些实施方案中,第一cas8亚基蛋白的n端通过第一连接子多肽与第一foki的c端共价连接,第一cas8亚基蛋白的c端通过第一连接子多肽与第一foki的n端共价连接,第二cas8亚基蛋白的n端通过第二连接子多肽与第二foki的c端共价连接,第二cas8亚基蛋白的c端通过第二连接子多肽与第二foki的n端共价连接,以及以上的组合。第一cas8亚基蛋白和第二cas8亚基蛋白中的每个可以包含具有不同序列的cas8亚基蛋白,或者第一和第二cas8亚基蛋白均可以包含相同的氨基酸序列。
[0034]
类似地,第一cse2亚基蛋白和第二cse2亚基蛋白中的每个可以包含不同的或相同的cse2亚基蛋白氨基酸序列,第一cas5亚基蛋白和第二cas5亚基蛋白中的每个可以包含不同的或相同的cas5亚基蛋白氨基酸序列,第一cas6亚基蛋白和第二cas6亚基蛋白中的每个可以包含不同的或相同的cas6亚基蛋白氨基酸序列,第一cas7亚基蛋白和第二cas7亚基蛋白中的每个可以包含不同的或相同的cas7亚基蛋白氨基酸序列,以及以上的组合。
[0035]
在优选的实施方案中,向导多核苷酸包含rna。
[0036]
在另外的实施方案中,本发明包括能够相对于野生型i型crispr cas3蛋白(“wtcas3蛋白”)减少沿dna的移动的改造的i型crispr cas3突变体蛋白(“mcas3蛋白”)。
[0037]
本发明还包括使用上述组合物进行细胞内的基因组编辑,以及制备上述组合物的方法。
[0038]
鉴于本文中的公开内容,本发明的其他实施方案对于本领域普通技术人员将易于变得显而易见。
[0039]
附图的简要说明
[0040]
图并非是成比例描绘的,它们也未描绘成一定比例。指示符的位置是近似的。
[0041]
图1a呈现了i型crispr-cas效应子复合体的一般化视图。图1b呈现了i型crispr-cas crrna的一般化视图。
[0042]
图2a、图2b和图2c呈现了具有结合至临近的间隔区序列的融合域的两种改造的i型crispr-cas效应子复合体的说明性实例。
[0043]
图3a和图3b呈现了环状排列的蛋白的实例。
[0044]
图4a、图4b、图5a、图5b、图6a、图6b、图6c、图7a、图7b、图8、图9、图10a和图10b示出了本发明的改造的i型crispr-cas效应子复合体的各种实例。
[0045]
图11a和图11b示出了底物通道的实例。
[0046]
图12a、图12b和图12c呈现了融合至cascade亚基蛋白的功能蛋白域被dcas9:natna复合体的位点定向募集的一般化视图。
[0047]
图13a、图13b、图14a、图14b和图14c示出了本发明的改造的i型crispr-cas效应子复合体的实例。
[0048]
图15a、图15b、图15c、图16a、图16b、图16c、图17a、图17b、图17c、图18a、图18b、图18c、图18d、图19a、图19b、图20a和图20b呈现了本发明的改造的i型crispr-cas效应子复合体的实例及其使用方法。
[0049]
图21a、图21b、图21c、图21d、图22a、图22b、图22c和图22d示出了本发明的使用包含活性核酸内切酶活性的cas3蛋白的实施方案。
[0050]
图23a、图23b、图23c、图23d、图23e、图24、图25、图26和图27呈现了各种cascade组分表达系统的示意图。
[0051]
图28、图29、图30、图31a、图31b、图32、图33a、图33b和图34呈现了与本发明的改造的cascade系统的基因组编辑相关的数据。
[0052]
图35示出了含有成对的向导rnas(grnas)的最小crispr阵列的实例。
[0053]
图36a、图36b、图36c和图36d呈现了与人类细胞中经由改造的i型crispr-cas复合体的基于rnp和质粒的递送的基因组编辑相关的数据。
[0054]
图37a、图37b、图37c、图37d、图37e、图37f和图37g呈现了与修复结果相关的数据。
[0055]
图38a、图38b和图38c呈现了与grnas和靶标dna之间的错配如何抑制改造的i型crispr-cas复合体的基因组编辑相关的数据。
[0056]
图39a、图39b、图39c和图39d呈现了与3种cascade同系物变体的pam选择性的扩大筛选相关的数据。
[0057]
图40a、图40b、图40c、图40d、图40e和图40f呈现了与改造的i型crispr-cas复合体的编辑效率的示例性变化相关的数据。
[0058]
图41a、图41b和图41c呈现了与3种cascade同系物变体的foki-cas8连接子长度和间隔区间距的扩大筛选相关的数据。
[0059]
图42a和图42b示出了以寡聚物为模板的pcr扩增的实例。
[0060]
图43呈现了基因组编辑百分比的数据,其显示为foki-cascade同系物变体和间隔区间距的函数。
[0061]
图44显示了ecocas3蛋白的功能域的线性示图和序列内制作的突变体的相对位置。
[0062]
图45a、图45b、图45c和图45d显示了与使用包含野生型或突变体ecocas3蛋白的ecocascade rnp复合体的基因组编辑相关的数据。
[0063]
图46a、图46b、图46c、图47a和图47b呈现了与dcas9-vp64/sgrna rnp复合体路障及其对ecocascade rnp复合体切割靶标的影响相关的数据。
[0064]
图48显示了cas3[d452a]/-ecocascade或mcas3[d452a]-ecocascade的示例性编辑数据。
[0065]
图49呈现了利用psecascade rnp复合体在8个trac靶标位点处进行基因组编辑的数据。
[0066]
通过引用并入本文
[0067]
在本说明书中引用的所有专利、出版物和专利申请都通过引用并入本文中,就好像每个单独的专利、出版物或专利申请都被明确地并且单独地指示为通过引用整体并入本文,用于所有目的。
[0068]
发明详述
blackswan(2008),isbn 978-8173716164。
[0072]
规律间隔成簇短回文重复序列(crispr)和相关的crispr相关蛋白(cas蛋白)构成了crispr-cas系统(参见,例如barrangou,r.,et al.,science 315:1709-1712(2007))。
[0073]
如本文所用,“cas蛋白”、“crispr-cas蛋白”和“crispr-cas亚基蛋白”及“cas亚基蛋白”,除非另外指出,否则均指第一类i型crispr-cas蛋白。通常,对于在本发明的方面中的用途,cas亚基蛋白能够与一种或多种同源的多核苷酸(最通常地,crrna)发生相互作用而形成i型效应子复合体(最通常地,rnp复合体)。
[0074]
随着时间的变化,以各种惯例对i-e型crispr
–
cas系统中的编码cascade的基因进行了命名,这在比较最新和较旧的文献时可能会成为一个混淆点。通常,本说明书使用的是如koonin,e.,et al.(curr.opin.microbiol.37:67-78(2017))中所示的的命名法,其中参考大肠杆菌k12操纵子中的基因顺序为:cas3、cas8、cas11、cas7、cas5、cas6、cas1和cas2。为简单起见,cas8e中的“e”限定词有时用于区分i型系统中不同亚型之间的cas8基因。野生型大肠杆菌i-e型crispr-cas的化学计量为cas5
1-cas6
1-cas7
6-cas8
1-cas11
2-grna1。
[0075]
然而,出于交叉引用的目的:cas8以前被称为cse1和casa,且也被称为“大亚基”;cas11以前被称为cse2和casb,且也被称为“小亚基”;cas7以前被称为cse4和casc;cas5以前被称为casd,且有时也被赋予了限定词cas5e;并且cas6以前被称为cse3和case,且通常被赋予限定词cas6e。
[0076]
表1列出了编码cas亚基蛋白的基因。
[0077][0078]
*如makarova,k.s.,et al.,nat.rev.microbiol.13:722-736(2015);koonin,e.v.,et al.,curr opin microbiol.37:67-78(2017)所定义的。
[0079]
pam序列通常被cas1亚基蛋白/cas2亚基蛋白复合体识别,其中活性pam感应位点与cas1亚基蛋白相关(参见,例如jackson,s.a.,et al.,science 356:356(6333)(2017))。cas1蛋白和cas2蛋白存在于绝大多数已知的crispr-cas系统中,且足够将间隔区插入crispr盒中(参见,例如yosef,i,et al.,nucleic acids res.40:5569
–
5576(2012))。这两种蛋白质形成了用于适应过程的复合体。cas1蛋白的核酸内切酶活性为间隔区整合所需要,而cas2蛋白似乎进行非酶促功能(参见,例如nunez,j.,et al.,nat struct mol biol.21:528
–
534(2014);richter,c.,et al.,plos one.2012;7:e49549)。cas1-cas2蛋白复合体代表了crispr-cas系统的高度保守的信息处理模块,其相对于系统的其余部分似乎是准自主的(参见,例如makarova,k.,et al.,methods mol.biol.1311:47-75(2015))。核
酸内切酶cas1蛋白是确保crispr系统保持对先前感染剂遭遇记忆的独特能力所必需的cas蛋白。
[0080]
术语“i型crispr-cas效应子复合体”、“i型crispr-cas核蛋白(np)复合体”、“cascade核蛋白(np)复合体”和“i型核蛋白(np)复合体”在本文中可互换使用,并且通常是指形成具有向导多核苷酸的复合体的cascade蛋白。当提及cascade np复合体的蛋白组分时,通常使用“cascade复合体”和“i型复合体”。术语“cascade rnp复合体”、“i型crispr-cas rnp复合体”和“i型rnp复合体”是指包含crrna相对于更一般的向导多核苷酸(即,如在cascade np复合体中)的cascade复合体。野生型i型crispr-cas效应子复合体的实例示出在图1a中。图1a自makarova,k.s.,et al.,(cell 168:946(2017);makarova,k.,et al.,nature reviews microbiology 13:722-736(2015))调整而来。图1a示出了结合为cascade复合体的6种cas7蛋白、cas5蛋白、cas8蛋白、两种cse2蛋白、cas6蛋白和crrna(图1a:cas7、cas5、cas8、cse2和cas6;cas6周围的虚线框是指其与crrna发卡的相互作用;crna示出为包括发卡的黑线)。复合体能够结合核酸靶序列。在wtcas3蛋白(图1a,虚线框所环绕的cas3)与复合体结合后,cascade复合体能够切割核酸靶序列。如表1所示,一些cas亚基蛋白的总数量在cascade复合体中可能存在差异。
[0081]“cas3”和“cas3蛋白”在本文可互换使用,是指i型crispr-cas3蛋白、其修饰和变体。i型crispr-cas效应子复合体结合与crrna向导互补的外来dna并募集cas3,cas3是靶标降解所需的反式作用核酸酶-解旋酶。cas3蛋白具有来自超家族2的解旋酶特征的基序,并包含dead/deah框区域和保守的c端域。cas3蛋白及其变体是本领域已知的(参见,例如westra,e.r.,et al.,mol.cell.46:595
–
605(2012);sinkunas,t.,et al.,embo j.30:1335-1342(2011);beloglazova,n.,et al.,embo j.30:4616-4627(2011);mulepati,s.,et al.,j.biol.chem.286:31896-31903(2011))。如本文所用的,术语“mcas3蛋白”是指相对于其相应的wtcas3蛋白包含一个或多个突变的cas3蛋白。mcas3蛋白包括但不限于:mcas3蛋白(例如,实施例23a、实施例23b和实施例23c)、dblmcas3蛋白(例如,实施例26a、实施例26b和实施例26c),以及dcas3*(不具有任何核酸酶活性和/或解旋酶活性的突变的cas3蛋白)。
[0082]
如本文所用的术语“核酸酶”是指酶,其能够切割如在双链(ds)核酸(例如,dsdna、基因组dna(gdna)、dsrna)、单链(ss)核酸(例如ssdna、rna)或杂交dsrna/dna中发现的诸如连接两个核苷酸的那些的磷酸二酯键。“核酸内切酶”通常可以影响其靶标分子中的ss-(裂口)或ds-缺口。dna核酸内切酶的一个实例是foki酶。“foki核酸内切酶”和“foki”在本文可互换使用,并且是指foki酶、foki同系物、foki酶的酶促活性域,以及foki酶的变体。foki二聚化通常是dna切割所需要的。foki的二聚物可以包含结合形成同型二聚体的两个单体亚基或者结合形成异型二聚体的两种不同的单体亚基(参见,例如bitinaite,j.,et al.,proc.natl.acad.sci.usa 95:10570-10575(1998);ramalingam,s.,et al.,j.mol.biol.405:630-641(2011))。foki变体的一个实例是guo,et al.(guo,j.,et al.,j.mol.biol.400:96-107(2010))所述的sharkey变体。另外的dna和rna核酸酶是本领域已知的。
[0083]
如本文所用的“crispr rna”、“crrna”和“向导rna”是指cas亚基蛋白能够与之相互作用而形成导引复合体优先结合多核苷酸中的核酸靶序列(相对于不包含核酸靶序列的
多核苷酸)的i型效应子复合体的一种或多种rnas。如本文所用的“向导”和“向导多核苷酸”是指包含核糖核苷酸碱基(例如,rna)和核糖的i型效应子复合体的多核苷酸组分,以及不同的组分及其组合,包括但不限于:脱氧核糖核苷酸碱基、核苷酸类似物、修饰的核苷酸、不同的含氮碱基、根本不同的核苷酸碱基、化学上不同的分子、碱基的混合物(例如,rna碱基、dna碱基和/或修饰的碱基)等,以及以上的组合,还有合成的骨架、天然存在的骨架、非天然存在的骨架、根本不同的骨架残基、化学上不同的残基或键、修饰的骨架、混合物(例如,骨架的核糖和脱氧核糖组分)等,以及以上的组合。本文描述了向导多核苷酸的一些实例。通过crrna间隔区与核酸靶序列结合的i型crispr-cas crrna的实例示出在图1b中。图1b自hochstrasser,m.l.,et al.,mol.cell 63:840-851(2016)调整而来。在图1b中,pam(图1b,104)与核酸靶序列结合,并示出了双链核酸的5
’
和3
’
链(图1b,垂直线代表氢键)。向导多核苷酸(图1b,106)通常包括5
’
柄区域(图1b,101)、包含种子区的间隔区(图1b,103),以及包含两个氢键重复区域的3
’
发卡(图1b,102);水平线代表氢键。本文讨论了与众多i型cascade同系物相关的pam序列。pam序列是相邻的前间隔区序列(图1b,105)。图1b示出了与核酸靶序列结合的cascade复合体间隔区(图1b,竖直线代表氢键)。图1b还示出了前间隔区(图1b,前间隔区)。间隔区可以包含约6至约56个核苷酸的crrna区域,其中间隔区与多核苷酸中的核酸靶序列互补。在i-e型crispr-cas系统中可以将间隔区长度改变为精细调节的cascade活性。cascade复合体可以掺入额外的cas7亚基,其中每6个核苷酸添加到crrna间隔区,并掺入额外的cse2亚基,其中每12个核苷酸添加到间隔区(参见,例如luo,m.l.,et al.,nucleic acids res.44(15):7385-7394(2016))。间隔区通常包含约32个至约36个核苷酸的区域。
[0084]
术语“间隔区”、“间隔区序列”和“核酸靶标结合序列”在本文可互换使用。
[0085]“靶标”、“靶序列”、“核酸靶序列”和“中靶序列”在本文可互换使用,指与cascade核蛋白复合体(例如,cascade rnp复合体)的向导的核酸靶标结合序列(例如,crrna的间隔区)完全或部分互补的核酸序列。通常,核酸靶标结合序列选择为与cascade核蛋白复合体的结合所指向的核酸靶序列100%互补;然而,为了减弱与核酸靶序列的结合,可以使用较低百分比的互补性。当靶标结合序列与靶序列100%互补时,“脱靶”序列结合是指cascade核蛋白复合体与和核酸靶标结合序列(间隔区)具有低于100%的互补性的核酸序列的结合。双链dna序列通常在一条链上包含核酸靶序列(图1b,与向导rna结合的部分氢)。“靶区”包含核酸靶序列。
[0086]
如本文中所使用的,“茎元件”或“茎结构”是指已知或预测形成双链区域(“茎元件”)的两条核酸链。“茎-环元件”或“茎-环结构”是指这样的茎结构,其中一条链的3
’
端序列通过通常为单链核苷酸的核苷酸序列(“茎-环元件核苷酸序列”)与第二链的5
’
端序列共价结合。在一些实施方案中,环元件包括长度为约3个至约20个核苷酸,优选长度为约4个至约10个核苷酸的环元件核苷酸序列。在优选的实施方案中,环元件核苷酸序列是不通过氢键形成相互作用而在环元件核苷酸序列内产生茎元件的未成对的核酸碱基的单链核苷酸序列。术语“发卡元件”在本文中也用于指茎环结构。这样的结构在本领域中是熟知的。碱基配对可能是准确的;然而,如本领域中已知的,茎元件不需要精确的碱基配对。因此,茎元件可包括一个或多个碱基错配或非成对的碱基。向导多核苷酸中的茎环结构的实例如图1b所示。
[0087]“连接子元件核苷酸序列”、“连接子核苷酸序列”和“连接子多核苷酸”在本文可互换使用,并且指共价附接至第一核酸序列(例如,5
’-
连接子核苷酸序列-第一核酸序列-3
’
)的一种或多种核苷酸的单链核酸序列或双链核酸序列。在一些实施方案中,连接子核苷酸序列连接两种不同的核酸序列而形成单一多核苷酸(例如,5
’-
第一核酸序列-连接子核苷酸序列-第二核酸序列-3
’
)。连接子核苷酸序列的其他实例包括但不限于:5
’-
第一核酸序列-连接子核苷酸序列-3
’
和5
’-
连接子核苷酸序列-第一核酸序列-连接子核苷酸序列-3
’
。在一些实施方案中,连接子元件核苷酸序列可以是不通过氢键形成彼此相互作用而在连接子元件核苷酸序列内产生二级结构(例如,茎-环结构)的未成对的核酸碱基的单链核苷酸序列。在一些实施方案中,两种连接子元件核苷酸序列可以通过两种连接子元件核苷酸序列之间的氢键彼此发生相互作用。在一些实施方案中,连接子多核苷酸编码“连接子多肽”。这样的连接子多核苷酸通常连接编码第一多肽的第一多核苷酸的3
’
端与编码第二多肽的第二多核苷酸的5
’
端,而形成编码包含n-第一多肽-连接子多肽-第二多肽-c的融合蛋白的单一多核苷酸。在本发明的一些实施方案中,可以通过连接子多肽串联连接超过两条多肽序列(例如,n-第一多肽-第一连接子多肽-第二多肽-第二连接子多肽-第三多肽-c)。“连接子多肽”、“连接子多肽序列”、“氨基酸连接子序列”和“连接子序列”在本文也可以互换使用。
[0088]
如本文所用,“连接核苷酸序列”是指共价连接第一核酸序列与第二核酸序列的单链核酸序列连接子序列。
[0089]
如本文所用,术语“间隔区间隔(interspacer)”、“间隔区间区(interspacer region)”和“间隔区间距”可互换使用,并且指第一核酸靶序列(例如,第一dna靶序列)的pam和第二核酸靶序列(例如,第二dna靶序列)的pam之间的通常在含pam(pam-in)方向上的距离,其中第一i型crispr-cas效应子复合体包含能够结合第一核酸靶序列的第一间隔区,并且第二i型crispr-cas效应子复合体包含能够结合第二核酸靶序列的第二间隔区。图2a、图2b和图2c呈现了包含融合蛋白的两种i型crispr-cas效应子复合体的说明性实例(图2a:“cascade1”,实线轮廓的框,包括“crrna1”;以及“cascade2”,虚线框,包括“crrna2”)(图2a,“fp1”和“fp2”表示为圆形部分;例如,fp1和fp可以是foki),其通过连接子多核苷酸与每个cascade复合体连接(图2a,“连接子1”和“连接子2”),其中crispr-cas效应子复合体与双链dna上的临近的核酸靶序列结合(图2a,“dsdna”,表示为成对的水平虚线)。指出了与每种核酸靶序列相关的pam序列(图2a,“pam1”,空心框,且“pam2”,空心框))。图2a示出了含pam(含pam/含pam)配置中的两个靶标位点之间的间隔区间隔(显示为图2a顶部的水平的双箭头线)。图2b示出了含pam/不含pam配置中的两个靶标位点之间的间隔区间隔(显示为图2b顶部的水平的双箭头线)。图2c示出了不含pam(不含pam/不含pam)配置中的两个靶标位点之间的间隔区间隔(显示为图2c顶部的水平的双箭头线)。图2a、图2b和图2c还示出了dsdna的两条链的分离。cascade复合体识别临近pam的dsdna靶序列。pam序列被cse1识别。crrna和互补靶标dna链之间的碱基配对导致产生具有取代的非互补的靶标dna链的r环(参见,例如beloglazova,n.,et al.,nucleic acids res.43:530
–
543(2015))。
[0090]
如本文所用,术语“同源的”是指相互作用的生物分子,如细胞表面受体(例如,趋化因子受体)及其配体(例如,在肿瘤细胞上或在肿瘤微环境中表达的趋化因子);位点定向多肽及其向导;能够位点定向结合与向导结合序列互补的核酸靶序列的位点定向多肽/向
978-0879694562(参见,例如appendix 1:structures of base pairs involving at least two hydrogen bonds,i.tinoco);principles of nucleic acid structure,w.saenger,springer international publishing ag(1988),isbn 978-0-387-90761-1;principles of nucleic acid structure,第一版,s.neidle,academic press(2007),isbn 978-01236950791)。
[0096]“连接(connect)”、“连接的(connected)”和“连接(connecting)”在本文可互换使用,并且指两个大分子(例如,多核苷酸、蛋白等)间的共价键或非共价键。
[0097]
如本文所用,术语“核酸序列”、“核苷酸序列”和“寡核苷酸”可互换使用,并且指核苷酸的聚合物形式。如本文所用,术语“多核苷酸”指具有一个5
’
端和一个3
’
端的核苷酸聚合物形式,并且可以包含一条或多条核酸序列。“环形多核苷酸”指在其5
’
端和其3
’
端间具有共价键,从而形成环形多核苷酸的多核苷酸。核苷酸可以是脱氧核糖核苷酸(dna)、核糖核苷酸(rna)、以上的类似物或以上的组合(例如,如以上向导多核苷酸的上下文中所述的),并且可以具有任何长度。多核苷酸可以进行任何功能,并可以具有各种二级和三级结构。该术语包括天然核苷酸和在碱基、糖和/或磷酸酯部分中修饰的核苷酸的已知类似物。特定核苷酸的类似物具有相同的碱基配对特异性(例如,a碱基对的类似物与t)。多核苷酸可以包含一个修饰的核苷酸或多个修饰的核苷酸。修饰的核苷酸的实例包括但不限于氟化核苷酸、甲基化核苷酸和核苷酸类似物。核苷酸结构可以在聚合物组装之前或之后进行修饰。在聚合后,多核苷酸可以另外经由例如与标记组分或靶标结合组分缀合而被修饰。核苷酸序列可以包含非核苷酸组分。还包括包含修饰的骨架残基或连接,即合成的、天然存在的和/或非天然存在,并且与参考多核苷酸(例如,dna或rna)具有相似的结合特性的核酸。此类类似物的实例包括但不限于硫代磷酸酯、氨基磷酸酯、甲基膦酸酯、手性甲基膦酸酯、2-o-甲基核糖核苷酸、肽-核酸(pnas)、锁核酸(lna
tm
)(exiqon,inc.,woburn,ma)核苷、乙二醇核酸、桥连核酸和吗啉代结构。
[0098]
肽-核酸(pnas)是核酸的合成同系物,其中多核苷酸磷酸糖骨架被柔性的伪肽聚合物取代,并且核碱基与聚合物连接。pnas具有以高亲和力和特异性与rna和dna的互补序列杂交的能力。
[0099]
在硫代磷酸酯核酸中,硫代磷酸酯(ps)键用多核苷酸磷酸酯骨架中的非桥接氧取代硫原子。这种修饰使核苷酸间连接抗核酸酶降解。在一些实施方案中,在多核苷酸序列的5
’
端或3
’
端的最后3-5个核苷酸之间引入硫代磷酸酯键,以抑制外切核酸酶降解。在整个寡核苷酸中放置硫代磷酸酯键也有助于减少核酸内切酶的降解。
[0100]
苏糖核酸(tna)是一种人工遗传聚合物。tna的骨架结构包括通过磷酸二酯键连接的重复的苏糖。tna聚合物抗核酸酶降解。tna可以通过碱基对氢键结合自组装成双链结构。
[0101]
可以通过使用“反向亚磷酰胺”将连接反向引入多核苷酸中(参见,例如www.ucalgary.ca/dnalab/synthesis/-modifications/linkages)。多核苷酸末端的3
’-3’
连接通过产生具有两个5
’-
oh端但缺少3
’-
oh端的寡核苷酸来稳定多核苷酸抗外切核酸酶的降解。通常,这样的多核苷酸在5
’-
oh位置具有亚磷酰胺基团,且在3
’-
oh位置具有二甲氧基三苯甲基(dmt)保护基团。通常,dmt保护基团在5
’-
oh上,且亚磷酰胺在3
’-
oh上。
[0102]
多核苷酸序列在本文中以常规的5
’-3’
方向显示,除非另外指出。
[0103]
如本文所用,“序列同一性”通常指使用具有不同权重参数的算法比较第一多核苷
酸或多肽与第二多核苷酸或多肽,而得出的核苷酸碱基或氨基酸的同一性百分比。可以通过利用万维网可获取的包括但不限于genbank(www.ncbi.nlm.nih.gov/genbank/)和embl-ebi(www.ebi.ac.uk)网站处的各种方法和计算机参数(例如,blast、cs-blast、psi-blast、fasta、hmmer、l-align等),使用序列比对确定两条多核苷酸或两种多肽之间的序列同一性。通常使用各种方法或计算机程序的标准默认参数计算两条多核苷酸或两种多肽序列之间的序列同一性。如本文所用两条多核苷酸或两种多肽之间的高度的序列同一性通常为约90%的同一性至100%的同一性,例如,约90%的同一性或更高,优选约95%的同一性或更高,更优选约98%的同一性或更高。如本文所用的两条多核苷酸或两种多肽之间的中度的序列同一性通常为约80%的同一性至约85%的同一性,例如,约80%的同一性或更高,优选约85%的同一性。如本文所用两条多核苷酸或两种多肽之间的低度的序列同一性通常为约50%的同一性至75%的同一性,例如,约50%的同一性,优选约60%的同一性,更优选约75%的同一性。例如,包含氨基酸取代的cas蛋白(例如,i-e型cse2、cas5、cas6、cas7和/或cas8)可以在其长度上与参考cas蛋白(例如,分别为野生型i-e型cse2、cas5、cas6、cas7和/或cas8)具有低度的序列同一性、中度的序列同一性或高度的序列同一性。作为另一个实例,向导多核苷酸在其长度上相比与参考cas蛋白复合的参考野生型向导多核苷酸(例如,与i-e型cse2、cas5、cas6、cas7和/或cas8形成复合体的向导多核苷酸)可以具有低度的序列同一性、中度的序列同一性或高度的序列同一性。
[0104]
如本文所用,“杂交(hybridization)”、“杂交(hybridize)”或“杂交(hybridizing)”是通过氢碱基配对组合两条互补的单链dna或rna分子以便形成单双链分子(dna/dna、dna/rna、rna/rna)的过程。杂交严格性通常由杂交温度和杂交缓冲液的盐浓度来确定;例如,高温和低盐提供了高严格性的杂交条件。不同杂交条件下的盐浓度范围和温度范围的实例如下:高严格性,约0.01m至约0.05m盐,杂交温度比tm低5℃-10℃;中等严格性,约0.16m至约0.33m盐,杂交温度比tm低20℃-29℃;以及低严格性,约0.33m至约0.82m盐,杂交温度比tm低40℃-48℃。通过本领域熟知的标准方法计算双链核酸序列的tm(参见,例如maniatis,t.,et al.,molecular cloning:a laboratory manual,cold spring harbor laboratory press:new york(1982);casey,j.,et al.,nucleic acids res.4:1539-1552(1977);bodkin,d.k.,et al.,j.virological methods 10:45-52(1985);wallace,r.b.,et al.,nucleic acids res.9:879-894(1981))。预估tm的算法预测工具也是广泛可用的。杂交的高严格条件通常是指与靶序列互补的多核苷酸主要与靶序列杂交而基本上不与非靶序列杂交的条件。通常,杂交条件是中等严格性,优选高严格性。
[0105]
如本文所用,“互补性”是指核酸序列与另一核酸序列(例如,通过典范的watson-crick碱基配对)形成氢键的能力。百分比互补性表示可以与第二核酸序列形成氢键的核酸序列中残基的百分比。如果两条核酸序列具有100%的互补性,则这两条序列是完全互补的,即,第一多核苷酸的所有连续残基与第二多核苷酸中的相同数量的连续残基氢键结合。
[0106]
如本文所用,“结合”是指大分子之间的非共价相互作用(例如,蛋白质与多核苷酸之间、多核苷酸与多核苷酸之间、蛋白质与蛋白质之间等)。这种非共价相互作用也称为“结合”或“相互作用”(例如,如果第一大分子与第二大分子相互作用,则第一大分子以非共价方式结合第二大分子)。结合相互作用的一些部分可以是序列特异性的(术语“序列特异性的结合”、“序列特异性结合”、“位点特异性的结合”和“位点特异性结合”在本文中可互换使
用)。如本文所用的序列特异性的结合通常指能够与i型crispr-cas亚基蛋白(例如,cse2、cas5、cas6、cas7和cas8)形成复合体,而优选相对于无核酸靶标结合序列(例如,dna靶标结合序列)的第二核酸序列(例如,第二dna序列),造成蛋白质结合包含核酸靶序列(例如,dna靶序列)的核酸序列(例如,dna序列)的一种或多种向导多核苷酸。结合相互作用的所有组分都不需要是序列特异性的,如蛋白质与dna骨架中的磷酸残基接触。结合相互作用的特征可以在于解离常数(kd)。“结合亲和力”是指结合相互作用的强度。结合亲和力增加与较低的kd相关。
[0107]
如本文所用,如果这样的复合体结合或切割多核苷酸内的核酸靶序列中的多核苷酸,则效应子复合体被说成是“靶向”多核苷酸。
[0108]
如本文所用,“双链断裂”(dsb)是指双链dna片段的两条链均被切断。在一些情况下,如果发生这种断裂,可以说一条链具有“粘性末端”,其中核苷酸暴露在外,且不与另一链上的核苷酸氢键结合。在其他情况下,可能会出现“钝末端”,其中两条链都保持与彼此完全碱基配对。
[0109]“供体多核苷酸”、“供体寡核苷酸”和“供体模板”在本文中可互换使用,并且可以是双链多核苷酸(例如,dna)、单链多核苷酸(例如,dna或rna)或以上的组合。供体多核苷酸可以包含在插入序列(例如,dna中的dsbs)两侧的同源臂。每侧上的同源臂的长度可以不同(例如,1-50个碱基、50-100个碱基、100-200个碱基、200-300个碱基、300-500个碱基、500-1000个碱基)。同源臂在长度上可以是对称的或非对称的。用于设计或构建供体多核苷酸的参数是本领域熟知的(参见,例如ran,f.,et al.,nature protocols 8:2281-2308(2013);smithies,o.,et al.,nature 317:230-234(1985);thomas,k.,et al.,cell 44:419-428(1986);wu,s.,et al.,nature protocols 3:1056-1076(2008);singer,b.,et al.,cell 31:25-33(1982);shen,p.,et al.,genetics 112:441-457(1986);watt,v.,et al.,proc.natl.acad.sci.usa 82:4768-4772(1985);sugawara,n.,et al.,j.mol.bio.12:563-575(1992);rubnitz,j.,et al.,j.mol.bio.4:2253-2258(1984);ayares,d.,et al.,proc.natl.acad.sci.usa 83:5199-5203(1986);liskay,r.,et al.,genetics 115:161-167(1987))。在一些实施方案中,供体多核苷酸包含嵌合的抗原受体(例如,car)。
[0110]
术语“嵌合的抗原受体”和“car”在本文中可互换使用,并且是指在实验室中创建的通常至少包含两种组分的多肽分子:胞外抗原识别域(也称为靶标结合域或胞外配体结合域)和胞内激活域(例如,包含一个或多个胞内信号传导域,并且通常包含一个或多个协同刺激信号传导域)。car还可以包含铰链域和跨膜域。典型的car多肽的结构如下:n端-胞外-[抗原识别域-铰链域]-跨膜-[跨膜域]-胞内-[胞内激活域]-c端;或n端-胞内
–
[胞内激活域]-跨膜
–
[跨膜域]
–
胞外
–
[抗原识别域-铰链域]-c端。
[0111]
胞外抗原识别域的实例包括用于与抗原结合的部分,并且包括但不限于单链免疫球蛋白可变片段(scfv)、抗原结合片段(fab;通常是结合抗原的抗体区域,且由每条重链和轻链的一个恒定域和一个可变域构成)、纳米抗体、骆驼科家族或鲨鱼来源的单链抗体、改造的蛋白结合支架(例如darpins和centyrins)或与其同源受体结合的天然配体。
[0112]
铰链域的实例包括但不限于可变长度(例如一个或多个氨基酸)的多肽铰链、cd8α的铰链区、cd28的铰链区、igg4的铰链区和以上的组合。
[0113]
跨膜域的实例包括但不限于来源于跨膜蛋白如cd8α、cd28、dap10、dap12、nkg2d和
以上的组合的跨膜区。
[0114]
胞内激活域的实例包括但不限于cd28、4-1bb、cd3ζ、ox40、2b4、dap10、dap12、截短和突变的信号传导域(例如,cd3ζ的三个itam域中的突变和截短)的胞内信号传导域,或其他胞内信号传导域,以及以上的组合。
[0115]
当胞外配体结合域与同源配体结合时,car的胞外信号传导域激活淋巴细胞(有关car-t细胞的描述,参见,例如brudno,j.,et al.,nature rev.clin.oncol.15:31-46(2018);maude,s.,et al.,n.engl.j.med.371:1507-1517(2014);sadelain,m.,et al.,cancer disc.3:388-398(2013);第7,446,190号美国专利;第8,399,645号美国专利)(有关car-nk细胞的描述,参见,例如rezvani,k.,et al.,mol.ther.,25:1769-1781(2017);siegler,e.,et al.,cell stem cell.23:160-161(2018);li,y.,et al.,cell stem cell.23:181-192(2018);lin,c.,et al.,biochim.biophys.acta.rev.cancer.1869:200-215(2018);hu,y.,et al.,acta.pharmacol.sin.39:167-176(2018);fang,f.,et al.,semin.immunol.31:37-54(2017);glienke,w.,et al.,front pharmacol.6:21(2015))。
[0116]
表2呈现了示例性细胞靶标和结合细胞靶标的scfvs/结合蛋白。这样的scfvs/结合蛋白或其部分可以掺入到car构建体中。
[0117]
[0118]
[0119][0120]
如本文所用,“同源定向修复”(hdr)是指在细胞内发生的dna修复,例如,在gdna中的dsb修复期间。hdr需要核苷酸序列同源性且使用供体或模板多核苷酸来修复序列,其中发生dsb(例如,在dna靶序列中)。供体多核苷酸通常具有需要的与dsb两侧的序列的序列同源性,使得供体多核苷酸可以用作用于修复的合适的模板。hdr导致遗传信息从例如供体多核苷酸传递至dna靶序列。如果供体多核苷酸序列与dna靶序列不同,并且供体多核苷酸的部分或所有被并入到dna靶序列中,则hdr可以造成dna靶序列的改变(例如,插入、缺失或突变)。在一些实施方案中,全部供体多核苷酸、一部分的供体多核苷酸或供体多核苷酸的拷贝被整合在dna靶序列的位点处。例如,供体多核苷酸可以用于dna靶序列中断裂的修复,其中修复造成来自供体多核苷酸的dna中断裂位点处或紧靠断裂的遗传信息的传递。因此,可以在dna靶序列处插入或拷贝新的遗传信息。
[0121]“基因组区域”是宿主细胞的基因组中的存在于核酸靶序列位点的任一侧上,或者可选地还包括一部分的核酸靶序列位点的染色体片段。供体多核苷酸的同源臂具有足够的同源性,可以与相应的基因组区域进行同源重组。在一些实施方案中,供体多核苷酸的同源臂与紧邻核酸靶序列位点两侧的基因组区域具有大量的序列同源性;公认的是,可以将同源臂设计成与远离核酸靶序列位点的基因组区域具有足够的同源性。
[0122]
如本文所用,“非同源末端连接”(nhej)指通过将断裂的一端直接连接到断裂的另一端来修复dna中的dsb,而无需供体多核苷酸。nhej是一种dna修复途径,可用于细胞修复dna而无需使用修复模板。在缺少供体多核苷酸的情况下,nhej通常会导致在dsb位点处随机插入或缺失核苷酸。
[0123]“微同源介导的末端连接”(mmej)是修复gdna中dsb的一种途径。mmej涉及dsb两侧的缺失,以及连接之前在断裂位点内部的微同源序列的对齐。mmej由基因限定,并且需要例如ctip、聚(adp-核糖)聚合酶1(parp1)、dna聚合酶θ(polθ)、dna连接酶1(lig 1)或dna连接酶3(lig 3)的活性。另外的遗传组分在本领域中是已知的(参见,例如sfeir,a.,et al.,trends in biochemical sciences 40:701-714(2015))。
[0124]
如本文所用,“dna修复”包括细胞机制借以修复对细胞中包含的dna分子的损伤的任何过程。修复的损伤可以包括单链断裂或dsbs。至少存在三种修复dsbs的机制:hdr、nhej和mmej。“dna修复”在本文中也用于指由人为或机器操纵而产生的dna修复,其中靶标位点被修饰,例如,通过插入、缺失或取代核苷酸,所有这些均代表了基因组编辑的形式。
[0125]
如本文所用,“重组”是指在两条多核苷酸之间进行遗传信息交换的过程。
[0126]
如本文所用,术语“调控序列”、“调控元件”和“控制元件”是可互换的,并且指多核苷酸靶标上游(5
’
非编码序列)、内部或下游(3
’
非翻译的序列)的待表达的多核苷酸序列。调控序列影响例如转录的时序;转录的量或水平;rna加工或稳定性;和/或相关结构核苷酸
序列的翻译。调控序列可包括活化剂结合序列、增强子、内含子、聚腺苷酸化识别序列、启动子、转录起始位点、阻抑剂结合序列、茎-环结构、翻译起始序列、内部核糖体进入位点(ires)、翻译前导序列、转录终止序列(例如,聚腺苷酸化信号和聚u序列)、翻译终止序列、引物结合位点等。
[0127]
调控元件包括指导许多类型的宿主细胞中的核苷酸序列的组成性的、可诱导的和可抑制的表达的那些,以及指导仅在某些宿主细胞(例如,组织特异性调控序列)中的核苷酸序列的表达的那些。在一些实施方案中,载体包含一个或多个pol iii启动子、一个或多个pol ii启动子、一个或多个pol i启动子,或以上的组合。pol iii启动子的实例包括但不限于u6和h1启动子。pol ii启动子的实例包括但不限于逆转录病毒鲁斯氏肉瘤病毒(rsv)ltr启动子(任选地与rsv增强子一起)、巨细胞病毒(cmv)启动子(任选地与cmv增强子一起;参见,例如boshart,m.,et al.,cell 41:521-530(1985))、sv40启动子、二氢叶酸还原酶启动子、β-肌动蛋白启动子、磷酸甘油激酶(pgk)启动子和ef1α启动子,以及改造的人工启动子(例如,mnd启动子和cag启动子)。本领域技术人员将理解,表达载体的设计可取决于诸如待转化的宿主细胞的选择、所需表达水平等因素。可以将载体引入到宿主细胞中从而产生如本文所述的由核酸序列编码的rna转录物、蛋白质或肽,包括融合蛋白或肽。
[0128]
如本文所用的“基因”是指包含外显子和相关的调控序列的多核苷酸序列。基因还可以包含内含子和/或非翻译区(utr)。
[0129]
如本文所用,术语“可操作地连接”是指与彼此处于功能关联的多核苷酸序列或氨基酸序列。例如,如果调控序列调控或有助于多核苷酸的转录调节,则调控序列(例如,启动子或增强子)与编码基因产物的多核苷酸“可操作地连接”。可操作地连接的调控元件通常与编码序列相邻。然而,如果与启动子相距不超过数千个碱基或更多,则增强子可以发挥功能。另外,多顺反子构建体可包括多个编码序列,其通过包括2a自切割肽、ires元件等而仅使用一个启动子。因此,一些调控元件可与多核苷酸序列可操作地连接,但与多核苷酸序列不相邻。类似地,翻译调控元件有助于从多核苷酸调节蛋白质表达。
[0130]
如本文所用,“表达”是指从dna模板转录多核苷酸,产生例如信使rna(mrna)或其他rna转录物(例如,非编码的,如结构性或支架rnas)。该术语还指转录的mrna被翻译成肽、多肽或蛋白质的过程。转录物和编码的多肽可以统称为“基因产物”。如果多核苷酸来源于gdna,则在真核细胞中表达可能包括剪接mrna。
[0131]“编码序列”或“编码”选定多肽的序列,是当置于适当的调控序列控制下时,体外或体内转录(在dna的情况下)和翻译(在mrna的情况下)成多肽的核酸分子。编码序列的边界由5'端处的起始密码子和3'端的翻译终止密码子决定。
[0132]
如本文所用,通过“人工转录活化剂(ata)”或“人工转录因子(atf)”,意指能够将rna聚合酶ii全酶募集至与其相关的基因,从而引起目标基因的异位表达的复合体。这样的活化剂至少包括两种组分:(1)直接识别同源核苷酸序列并能够结合这些序列的催化失活的多核苷酸结合域,或者导向这样的序列以用于结合的多核苷酸结合域(例如,包含如本文所述的核酸结合域和向导的核蛋白复合体);以及(2)与构成了上调转录的转录机制的各种蛋白相互作用的激活域(也称为“效应子域”)。
[0133]
通过“催化失活的多核苷酸结合域”,意指结合但不切割被结合域结合的核酸靶标位点的分子。在本文中详细描述了此类域的代表性实例。
[0134]
如本文所用,术语“调节”是指功能的数量、程度或量的变化。例如,本文公开的i型crispr核蛋白复合体可以通过在启动子或转录起始位点或调控位点处或附近与核酸靶序列结合来调节启动子序列的活性。根据结合后发生的作用,i型crispr核蛋白复合体可以诱导、增强、压抑或抑制与启动子序列可操作地连接的基因的转录。因此,基因表达的“调节”包括基因激活和基因抑制。
[0135]
可以通过确定直接或间接受靶标基因表达影响的任何特征来分析调节。这些特征包括例如,rna或蛋白质水平、蛋白质活性、产物水平、基因的表达或报告基因的活性水平的变化。因此,术语“调节基因的表达”、“抑制基因的表达”和“激活基因的表达”可以指i型crispr核蛋白复合体改变、激活或抑制基因转录的能力。
[0136]
功能(例如,酶促功能)可以被上调(例如,增加、增强、扩增或加强功能)或下调(例如,降低、减弱,减小或减少功能)。在一个实施方案中,可以相对于相应的wtcas3蛋白上调或下调mcas3蛋白与单链dna(ssdna)结合或者通过mcas3蛋白atp结合/水解。
[0137]
如本文所用的“载体”和“质粒”,是指将遗传物质引入细胞内的多核苷酸媒介物。向量可以是线性的或环形的。载体可以包含复制序列,该复制序列能够实现载体在合适的宿主细胞中的复制(例如,复制起点)。在转化合适的宿主后,载体可以独立于宿主基因组复制和发挥功能,也可以整合到宿主基因组中。除其他外,载体设计取决于载体的预期用途和宿主细胞,并且用于特定用途和宿主细胞的本发明载体的设计在本领域技术水平内。载体的四种主要类型是质粒、病毒载体、粘粒和人工染色体。通常,载体包含复制起点、多克隆位点和/或可选择标记。表达载体通常包含表达盒。通过“重组病毒”,意指已经例如将异源核酸构建体添加或插入病毒基因组或其一部分而进行遗传改变的病毒。
[0138]
如本文所用,“表达盒”是指使用重组方法或通过合成手段产生,并包含可操作地连接至所选多核苷酸以促进所选多核苷酸在宿主细胞中表达的调控序列的多核苷酸构建体。例如,调控序列可以促进选定的多核苷酸在宿主细胞中的转录,或选定的多核苷酸在宿主细胞中的转录和翻译。例如,可以将表达盒整合到宿主细胞的基因组中,也可以存在于载体中以形成表达载体。
[0139]
如本文所用,“靶向载体”是重组dna构建体,其通常包含与gdna同源的特制dna臂,位于靶标基因或核酸靶序列(例如,dsb)的两侧。靶向载体包含供体多核苷酸。靶标基因的元件可以多种方式进行修饰,包括缺失和/或插入。有缺陷的靶标基因可以用功能靶标基因代替,或者可选地可以将功能基因敲出。靶向载体的供体多核苷酸包含选择盒,该选择盒包含引入靶标基因中的可选择标记。邻近或位于靶标基因内的靶向区域(包含核酸靶序列)可用于实现基因表达的调控。
[0140]
如本文所用,术语“在
…
之间”包括给定范围内的终点值(例如,在1-50个核苷酸之间的长度包括1个核苷酸和50个核苷酸;在5个氨基酸至50个氨基酸之间的长度包括5个氨基酸和50个氨基酸)。
[0141]
如本文所用,术语“氨基酸”(aa)是指天然的和合成的(非天然的)氨基酸,包括氨基酸类似物、修饰的氨基酸、模拟肽、甘氨酸,以及d或l光学异构体。
[0142]
如本文所用,术语“肽”、“多肽”、“蛋白”和“亚基蛋白”是可互换的,并且指氨基酸的聚合物。多肽可以具有任何长度。其可以是分枝的或线性的,其可以插入有非氨基酸,并且其可以包含修饰的氨基酸。该术语也指已经通过例如乙酰化、二硫键形成、糖基化、脂质
化、磷酸化、聚乙二醇化、生物素化、交联和/或缀合(例如,利用标记组分或配体)修饰的氨基酸聚合物。除非另有说明,否则本文中的多肽序列以常规的n端至c端方向显示。
[0143]
可以使用分子生物学领域中的常规技术制备多肽和多核苷酸(参见,例如上面列出的标准文本)。此外,基本上任何的多肽或多核苷酸都可从商业来源获得。
[0144]
如本文所用的术语“融合蛋白”和“嵌合蛋白”是指通过连接非天然一起存在于单一蛋白中的两个或更多个蛋白、蛋白域、蛋白片段或环形排列的多肽而产生的单一蛋白。在一些实施方案中,连接子多核苷酸可用于将第一蛋白、蛋白域或蛋白片段或循环排列的多肽连接至第二蛋白、蛋白域、蛋白片段或循环排列的多肽。例如,融合蛋白可以包含i型crispr-cas蛋白(例如,cas8、cas3)和来自另一蛋白的功能域(例如,foki;参见,例如第9,885,026号美国专利)。进行修饰以在融合蛋白中包括这样的域可以对改造的i型crispr-cas蛋白赋予另外的活性。这样的活性可以包括修饰与核酸靶序列(例如,组蛋白)相关的多肽的核酸酶活性、甲基转移酶活性、脱甲基酶活性、dna修复活性、dna损伤活性、脱氨活性、歧化酶活性、烷化活性、脱嘌呤活性、氧化活性、嘧啶二聚体形成活性、整合酶活性、转座酶活性、重组酶活性、聚合酶活性、连接酶活性、解旋酶活性、光裂合酶活性、糖基化酶活性、乙酰转移酶活性、脱乙酰酶活性、激酶活性、磷酸酶活性、泛素连接酶活性、去泛素化活性、腺苷酰化活性、脱腺苷化活性、sumo化活性、脱sumo化活性、核糖基化活性、脱核糖基化活性,和/或豆蔻酰化活性或脱豆蔻酰化活性。
[0145]
在一些实施方案中,融合蛋白可以包含表位标签(例如,组氨酸标签、ha标签、(sigma aldrich,st.louis,mo)标签、myc标签、核定位信号(nls)标签,suntag)、报告子蛋白序列(例如,谷胱甘肽-s-转移酶、β-半乳糖苷酶、荧光素酶、绿色荧光蛋白、青色荧光蛋白、黄色荧光蛋白),和/或核酸序列结合域(例如,dna结合域或rna结合域)。
[0146]
融合蛋白还可以包含活化剂域(例如,热休克转录因子nfkb活化剂)或阻抑剂域(例如,krab域)。如lupo,a.,et al.,current genomics 14:268-278(2013)所述的,krab域是有效的转录抑制模块,且位于大多数c2h2锌指蛋白的氨基端序列中(参见,例如margolin,j.,et al.,proc.natl.acad.sci.usa 91:4509-4513(1994);witzgall,r.,et al.,proc.natl.acad.sci.usa 91:4514-4518(1994))。krab域通常经由蛋白-蛋白相互作用与协阻抑物蛋白和/或转录因子结合,造成与krab锌指蛋白(krab-zfps)结合的基因的转录抑制(参见,例如friedman,j.r.,et al.,genes&development 10:2067-2678(1996))。在一些实施方案中,连接子核酸序列被用于连接两个或更多个蛋白、蛋白域或蛋白片段。
[0147]
如本文所用,“cascadea”(cascade激活)是一种crispr方法或系统,其中所述方法或系统激活与cascade rnp复合体的靶标核酸序列的位点相关的基因的表达。在一些实施方案中,cascade复合体的一种或多种蛋白被融合至效应子域(例如,vp16或vp64),并且包含融合和向导多核苷酸的cascade rnp复合体被用于募集内源转录因子。在一些实施方案中,向导多核苷酸可以经5
’
或3
’
融合至核苷酸效应子域,如也募集转录因子的ms2结合rna。
[0148]
如本文所用,“cascadei”(cascade抑制)是一种crispr方法或系统,其中所述crispr方法或系统下调与cascade rnp复合体的靶标核酸序列的位点相关的基因的表达(即,cascade rnp复合体被用于下调基因的表达)。对于内源抑制因子的募集,cascade复合体中的一种或多种蛋白质通常被融合至效应子域(例如,krab)。在一些实施方案中,向导多核苷酸可以经5
’
或3
’
融合至也募集内源转录抑制效应子蛋白的核苷酸效应子域。
[0149]
如本文所用的“部分”是指一部分的分子。部分可以是功能基团,也可以描述具有多个功能基团(例如,共用共同的结构方面)的一部分分子。术语“部分”和“功能基团”在本文中通常可互换使用;然而,“功能基团”可以更具体地指包括一些常见化学行为的一部分分子。“部分”通常用作结构描述。在一些实施方案中,5'端、3'端或5'端和3'端(例如,第一茎元件中的非天然的5'端和/或非天然的3'端)可包含一个或多个部分。
[0150]
如本文所用,“过继细胞”是指可以进行遗传修饰以用于细胞疗法的细胞,如用于治疗癌症和/或预防移植物抗宿主病(gvhd)以及细胞疗法的其他不良副作用,诸如但不限于细胞因子风暴、所给予的遗传改良物质的致癌性转化、神经性病症等。过继细胞包括但不限于干细胞、诱导性多能干细胞(ipscs)、脐带血干细胞、淋巴细胞、巨噬细胞、红细胞、成纤维细胞、内皮细胞、上皮细胞和胰腺前体细胞。
[0151]
如本文所用,“细胞疗法”是指利用遗传改良的细胞治疗疾病或病症。可以使用本文描述的方法来引入遗传修饰,如包括病毒载体、核转染、基因枪递送、超声处理、细胞挤压、脂转染或使用其他化学物质、细胞穿透肽等的方法。
[0152]
如本文所用,“过继细胞疗法(act)”是指使用来源于返回至该患者的特定患者(自体细胞疗法)或第三方供体(异体细胞疗法)的遗传改良的过继细胞来治疗患者的疗法。acts包括但不限于骨髓移植、干细胞移植、t细胞疗法、car-t细胞疗法和自然杀伤(nk)细胞疗法。
[0153]
如本文所用,“淋巴细胞”是指作为脊椎动物免疫系统的一部分的白细胞(白血细胞)。术语“淋巴细胞”还包括造血干细胞或产生淋巴样细胞的诱导多能干细胞(ipsc)。淋巴细胞包括用于细胞介导的细胞毒性适应性免疫的t细胞,如cd4+和/或cd8+细胞毒性t细胞;α/βt细胞和γ/δt细胞;调节性t细胞,如treg细胞;在细胞介导的细胞毒性先天免疫中起作用的nk细胞;用于体液、抗体驱动的适应性免疫的b细胞;nk/t细胞;细胞因子诱导的杀伤细胞(cik细胞);以及抗原呈递细胞(apcs),如树突细胞。淋巴细胞可以是哺乳动物细胞,如人(智人;h.sapiens)细胞。术语“淋巴细胞”还包括经遗传改良的t细胞和nk细胞,其被修饰以在t或nk细胞表面(car-t细胞和car-nk细胞)上产生嵌合抗原受体(cars)。这些car-t细胞识别特定的可溶性抗原或靶标细胞表面,如肿瘤细胞表面,或肿瘤微环境中的细胞上的抗原。
[0154]
如本文所用,术语“淋巴细胞”还包括t细胞受体改造的t细胞(tcrs),,其经遗传改良以表达一种或多种特定的天然存在的或改造的t细胞受体,后者可以识别主要组织相容性复合体(mhc)所呈递的靶标细胞的蛋白或(糖)脂质抗原。这些抗原的小片段,如肽或脂肪酸,被穿梭到靶标细胞表面,并呈递给作为mhc一部分的t细胞受体。t细胞受体与载有抗原的mhcs的结合激活淋巴细胞。
[0155]
当淋巴细胞通过其细胞表面上的抗原特异性受体触发时,就会发生淋巴细胞活化。这导致细胞增殖并分化为特异的效应子淋巴细胞。这种“激活的”淋巴细胞通常以淋巴细胞表面上的一组受体为特征。活化的t细胞的表面标记包括cd3、cd4、cd8、pd1、il2r等。活化的细胞毒性淋巴细胞可以在结合靶标细胞表面上的同源受体后杀伤靶标细胞。
[0156]
如本文所用,术语“淋巴细胞”也包括肿瘤浸润淋巴细胞(tils)。tils是已经穿透了肿瘤中及其周围环境(“肿瘤微环境”)的免疫细胞。tils通常是从肿瘤细胞和肿瘤微环境中分离出来的,并在体外挑选了针对肿瘤抗原的高反应性。tils在克服体内存在的耐受性
影响的条件下体外生长,然后引入对象内进行治疗。
[0157]
t细胞通常呈现许多亚型,如“幼稚t细胞”(tn)、“干细胞记忆t细胞”(tscm)、“中央记忆t细胞”(tcm)“效应子记忆t细胞”(tem)、“效应子t细胞”(teff)和“调节性t细胞”(treg)。每个t细胞亚组的特征是一组细胞表面标记。
[0158]
如本文所用的术语“亲和标签”通常是指增加一个大分子对另一个大分子的结合亲和力的一个或多个部分,例如,以促进形成改造的i型crispr-cas核蛋白复合体。在一些实施方案中,亲和标签可用于增加一种cas亚基蛋白对另一种cas亚基蛋白(例如,第一cas7蛋白对第二cas7蛋白)的结合亲和力。在一些实施方案中,亲和标签可用于增加一种或多种cas亚基蛋白对同源的向导多核苷酸的结合亲和力。本发明的一些实施方案将一种或多种亲和标签引入cas亚基蛋白质序列的n端、cas亚基蛋白序列的c端、位于cas亚基蛋白质序列的n端和c端之间的位置,或以上的组合。在本发明的一些实施方案中,一种或多种向导多核苷酸包含亲和标签,该亲和标签增加了向导多核苷酸与一种或多种cas亚基蛋白质的结合亲和力。在2014年10月23日公开的第2014-0315985号美国公开专利申请中公开了各种各样的亲和标签。配体和配体结合部分是成对的亲和标签。
[0159]
如本文所用,“交联”是将一个聚合物链(例如,多核苷酸或多肽)与另一个连接的键。这样的键可以是共价键或离子键。在一些实施方案中,一个多核苷酸可以通过交连多核苷酸而与另一个多核苷酸结合。在其他实施方案中,可以将多核苷酸与多肽交联。在另外的实施方案中,可以将多肽与多肽交联。
[0160]
如本文所用,术语“交联部分”通常是指适合在两个大分子之间提供交联的部分。交联部分是亲和标签的另一个实例。
[0161]
如本文所用,“宿主细胞”通常是指生物细胞。细胞是生物的基本结构、功能和/或生物学单元。细胞可以源自具有一个或多个细胞的任何生物。宿主细胞的实例包括但不限于原核细胞、真核细胞、细菌细胞、古菌细胞、单细胞真核生物的细胞、真核生物的细胞、原生动物细胞、来自植物的细胞、藻类细胞(例如,布朗葡萄藻(botryococcus braunii)、莱茵衣藻(chlamydomonas reinhardtii)、迦得拟微绿球藻(nanonochloropsis gaditana)、蛋白核小球藻(chlorella pyrenoidosa)、展枝马尾藻圆干变种(sargassum patens c.agardh)等)、海藻(例如,巨藻)、真菌细胞(例如,酵母细胞或来自蘑菇的细胞)、动物细胞、来自无脊椎动物(例如,果蝇、刺胞动物、棘皮动物、线虫等)的细胞、来自包括哺乳动物在内的脊椎动物(例如,猪、牛、山羊、绵羊、啮齿动物、大鼠、小鼠、非人的灵长类动物、人类等)的细胞。此外,宿主细胞可以是干细胞或祖细胞,以及免疫细胞,如本文所述的任何免疫细胞。宿主细胞可以是人类细胞.。在一些实施方案中,人类细胞在人体外部。在一些实施方案中,对活的生物体(例如人体)的体细胞进行离体操作(即在活体外)。离体通常是指从活体(例如,人体)采集器官、细胞或组织以进行治疗或手术,然后再返回活体的医疗程序。
[0162]
如本文所用,“干细胞”是指具有自我更新能力,即经历许多次循环的细胞分裂同时又保持未分化状态的能力的细胞。干细胞可以是全能的、多能的、专能的、寡能的或单能的。干细胞可以是胚胎、胎儿、羊膜、成年或诱导的多能干细胞。
[0163]
如本文所用,“诱导的多能干细胞”是指从非多能细胞,通常是体细胞人工衍生而来的一类多能干细胞。在一些实施方案中,体细胞是人类体细胞。体细胞的实例包括但不限于真皮成纤维细胞、骨髓来源的间充质细胞、心肌细胞、角质形成细胞、肝细胞、胃细胞、神
经干细胞、肺细胞、肾细胞、脾细胞和胰腺细胞。体细胞的其他实例包括免疫系统的细胞,包括但不限于b细胞、树突状细胞、粒细胞、先天淋巴样细胞、巨核细胞、单核细胞/巨噬细胞、髓样来源的抑制细胞、自然杀伤(nk)细胞、t细胞、胸腺细胞和造血干细胞。
[0164]
如本文所用,“造血干细胞”是指具有分化为造血细胞如淋巴细胞的能力的未分化细胞。
[0165]
如本文所用,“植物”是指整株植物、植物器官、植物组织、种质、种子、植物细胞及其后代。植物细胞包括但不限于来自种子、悬浮培养物、胚、分生组织区域、愈伤组织、叶、根、枝、配子体、孢子体、花粉和小孢子的细胞。植物部位包括分化和未分化的组织,包括但不限于根、茎、枝、叶、花粉、种子、肿瘤组织以及各种形式的细胞和培养物(例如,单细胞、原生质体、胚和愈伤组织)。植物组织可以在植物中或在植物器官、组织或细胞培养物中。“植物器官”是指构成植物的形态和功能上不同的部分的植物组织或一组组织。
[0166]
术语“对象”、“个体”或“患者”在本文中可互换使用,并且指脊索动物门的任何成员,包括但不限于人类和其他灵长类,包括非人的灵长类,如恒河猴、黑猩猩和其他猴子和猿类物种;农场动物,如牛、绵羊、猪、山羊和马;家养哺乳动物,如狗和猫;实验室动物,包括兔子、小鼠、大鼠和豚鼠;鸟类,包括家养的、野生的和游戏性鸟类,如鸡、火鸡和其他鸡形鸟类、鸭子和鹅;等等。该术语不表示特定的年龄或性别。因此,该术语包括成年、年轻和新生个体以及雄性和雌性。在一些实施方案中,宿主细胞来源于对象(例如,淋巴细胞、干细胞、祖细胞或组织特异性细胞)。在一些实施方案中,对象是非人对象。在一些实施方案中,对象是人类(智人)对象。
[0167]
术语“有效量”或“治疗有效量”的组合物或试剂,如本文提供的遗传改造的过继细胞,是指足以提供所需反应,如预防或消除与同种异体过继细胞疗法有关的一种或多种有害副作用的组合物或试剂的量。此类反应将取决于特定目标疾病。例如,在使用过继细胞疗法治疗癌症的患者中,所需的反应包括但不限于治疗或预防gvhd、宿主抗移植物排斥、细胞因子释放综合征(crs)、细胞因子风暴,以及减少给予的遗传改良细胞的致癌转化的影响。所需的确切量将因对象而异,这取决于对象的种类、年龄和一般状况、所治疗病况的严重程度、所用的特定改良淋巴细胞、给药方式等。在任何个别情况下,本领域普通技术人员可以使用常规实验确定适当的“有效”量。
[0168]“治疗(treatment)”或“治疗(treating)”特定疾病,如癌性病况或gvhd,包括:(1)预防疾病,例如,预防疾病的发展或在可能易患该疾病,但尚未经历或未显示该疾病的症状的对象中使疾病以较低的强度发生;(2)抑制疾病,例如,降低发展速度,阻止发展或逆转疾病状态;和/或(3)减轻疾病的症状,例如,减少对象经历的症状的数量。
[0169]
如本文所用,通过“基因编辑”或“基因组编辑”意指导致基因修饰的一类基因工程,如位于细胞基因组中的特定位点处的核苷酸序列,或甚至单个碱基的插入、缺失或替换。该术语包括但不限于如本文所定义的异源基因表达、基因或启动子插入或缺失、核酸突变和破坏性遗传修饰。
[0170]
通过“表位”意指分子上特定b细胞和t细胞对其响应的位点。在表位独特性的空间配置中,表位可以包含3个或更多个氨基酸。通常,表位由至少五个这样的氨基酸组成,且更通常地,由至少8-10个这样的氨基酸组成。确定氨基酸的空间配置的方法是本领域已知的,并且包括例如x射线晶体学、电子显微镜和二维核磁共振。此外,使用本领域熟知的技术,如
通过使用疏水性研究和通过位点定向血清学,可以容易地完成给定蛋白质中表位的鉴定。
[0171]“模拟表位”是模拟表位结构的大分子,如肽。由于这种特性,它会导致类似于表位引发的抗体反应。给定表位抗原的抗体将识别模拟该表位的模拟表位。模拟表位通常是通过生物淘选从噬菌体展示库获得的。
[0172]“抗体”意指一种“识别”,即特异性结合在诸如配体结合域之类的多肽中呈现的目标表位的分子。通过“特异性结合”,意指抗体以“锁和钥匙”型相互作用与表位相互作用,以在抗原和抗体之间形成复合体。如本文所用,术语“抗体”包括从单克隆制剂获得的抗体,以及以下各项:杂交(嵌合)抗体分子;f(ab
’
)2和f(ab)片段;fv分子(非共价异型二聚体;单链fv分子(scfv);二聚和三聚抗体片段构建体;小抗体;人源化抗体分子;单链抗体;纳米抗体(ablynx n.v.,zwijnaarde,belgium)抗体;以及从这些分子获得的任何功能片段,其中这样的片段保留了亲本抗体分子的免疫学结合特性。抗体可以来源于不同的物种,如人类、小鼠、大鼠、兔、骆驼、鸡等。然后可以通过体外技术,如通过噬菌体展示和酵母展示进一步获得抗体和抗体部分。可以从人血浆、人b细胞克隆、小鼠、大鼠、兔、鸡等获得完全人源化的抗体,这些抗体具有改造的人源化b细胞库。然后可以通过亲和力成熟和其他方法,如岩藻糖基化或igg fc工程进一步修饰抗体。
[0173]
如本文所用,术语“单克隆抗体”是指具有同质抗体群的抗体组合物。该术语不限于关于抗体的种类或来源,也不旨在受其制备方式的限制。该术语包括完整的免疫球蛋白以及诸如fab、f(ab
’
)2、fv和其他片段等片段,以及嵌合和人源化均质抗体群,它们均显示本发明亲本单克隆抗体分子的免疫结合特性。
[0174]“抗体依赖性细胞介导的细胞毒性(adcc)”也称为“抗体依赖性细胞细胞毒性”,是指当膜表面配体结合域已被特定抗体结合时,免疫系统的效应子细胞借以主动裂解靶标细胞,如过继细胞的机制。效应子细胞通常是自然杀伤(nk)细胞。然而,巨噬细胞、嗜中性粒细胞和嗜酸性粒细胞也可以介导adcc。adcc独立于补体依赖性细胞毒性(cdc),后者也可通过破坏膜来裂解靶标,而无需抗体或免疫系统细胞的参与。
[0175]
如本文所用,“转化”是指将外源多核苷酸插入宿主细胞,而与用于插入的方法无关。例如,可以通过直接摄取、转染、感染等来进行转化。外源多核苷酸可以维持为非整合载体,例如附加体,或者可选地,可以整合到宿主基因组中。如本文所用,“转基因生物体”是指含有遗传物质的生物体,其中已人工引入了来自不相关生物体的dna。该术语包括转基因生物体的后代(任何世代),只要该后代具有遗传修饰。在一些实施方案中,转基因生物体是非人的转基因生物体。
[0176]
如本文所用,“分离的”可以指通过人为干预而存在于其天然环境之外并因此不是天然产物的分子(例如,多核苷酸或多肽)。当提及多肽时,分离的意指所示分子与在天然下与该分子一起发现的完整生物体分离且不连续,或在基本上没有其他相同类型的生物大分子的情况下出现。关于多核苷酸的术语“分离的”是全部或部分缺乏天然下通常与其相关的序列的核酸分子;或者序列,当其在天然下存在时,但是具有与其相关的异源序列;或者与染色体分离的分子。
[0177]
如本文所用,术语“纯化的”优选意指存在根据重量至少75%,更优选根据重量至少85%,仍然更优选根据重量至少95%,且最优选根据重量至少98%的相同分子。
[0178]
如本文所用,“底物通道”是指反应物从一种酶促反应直接转移到另一种酶促反应
中,而没有首先扩散到整体环境中(参见,例如,wheeldon,i.,et al.,nat.chem.8:299-309(2016))。这些酶促步骤的中间体与整体溶液不平衡,这使得酶促过程的效率和产率得以提高。通常,在天然发生的代谢过程中,酶已发展出共定位和组装成受控聚集体的方式。
[0179]
如本文所用,“底物通道元件”是指代谢途径的组成部分。在一些实施方案中,底物通道元件是一种催化化学反应的酶。
[0180]
如本文所用,“底物通道复合体”是指经一些方式一起共定位的多个底物通道元件。
[0181]
如本文所用,“rna支架”是指肽可以用作结合底物的rna分子。
[0182]
本文提供的数据证明了cascade组分和核酸酶域(例如,二聚化依赖性的非特异性foki核酸酶域;参见,例如urnov,f.d.,et al.,nature reviews genetics 11:636
–
646(2010);joung,j.k.,et al.,nat.rev.mol.cell biol.14:49
–
55(2013);guilinger,j.p.,et al.,nat.biotechnol.32:577
–
582(2014);tsai,s.q.,et al.,nat.biotechnol.32:569
–
576(2014))之间的融合介导人类细胞中利用i型系统的有效的可编程rna引导的基因编辑。数据证明改造的i型crispr-cas系统(例如,包括foki-cascade组分融合)可被直接转染为完整的核糖核蛋白(rnp)复合体或者经由单个质粒编码的组分的递送在细胞中组装。如本文中所陈述,所有crispr相关的(cas)基因都被组装在单个多顺反子载体上,产生简化的双组分cas蛋白引导的rna表达系统。另外,核酸酶(例如,foki)/cascade组分连接子序列的长度/组合物设计和适当的dna几何学设置,以及选择性cascade同系物选择,提供了具有多达约50%的编辑效率的改造的i型crispr-cas复合体。确定了涉及dna靶向期间的pam需求和错配敏感性的改造的i型crispr-cas系统(例如,包括foki
–
cascade组分融合蛋白)的关键特征。
[0183]
在第一个方面,本发明涉及编码包括但不限于cascade亚基蛋白和cascade向导多核苷酸的cascade组分的改造的多核苷酸。
[0184]
在一个实施方案中,本发明涉及编码来源于cascade i-e型系统的cascade组分的改造的多核苷酸。在实施例1中呈现了包含cascade蛋白和cascade crrnas的示例性多核苷酸构建体。实施例1、表15和seq id no:1至seq id no:20呈现了编码具体地来自大肠杆菌菌株k-12 mg1655的i-e型cascade的5种亚基蛋白的基因的多核苷酸dna序列,以及所得蛋白质组分的氨基酸序列。多核苷酸序列来源于大肠杆菌gdna,并且具体地针对在大肠杆菌中表达进行了密码子优化,和/或具体地针对在真核细胞(例如,人类细胞)中表达进行了密码子优化。当该多核苷酸被转录到前体crrna中并通过cascade rna核酸内切酶处理时,产生了用作向导rna的成熟的crrna,以靶向基因组中的互补dna序列。最小crispr阵列包含在示例性间隔区序列两侧的两条重复序列(在实施例1中呈现的crispr阵列序列中画有下划线的),其代表crrna的向导部分。通过cascade核酸内切酶加工rna产生了在5
’
和3
’
端于向导序列两侧的具有重复序列的crrna。本领域普通技术人员参考本说明书和实施例的教导,可以选择适当的间隔区序列来靶标结合cascade复合体至选择的靶序列(例如,在gdna中)。
[0185]
可以根据本说明书的指导,并使用生物信息学工具如blast和psi-blast鉴定和设计编码来自另外的细菌和古菌物种的cascade组分的多核苷酸序列,以定位例如来自大肠杆菌菌株k-12mg1655的cascade亚基基因的同系物,然后检查cascade基因的两侧的基因组邻近性,从而定位和鉴定剩余cascade亚基蛋白的基因(参见,例如实施例14a、实施例14b、
实施例15a和实施例15b)。由于cascade基因作为保守的操纵子同时出现,因此它们通常以一致的顺序排列在相同的i型亚型中,从而促进了它们的鉴定和选择,以进行后续分析和实验。例如,可以通过定位cas8同系物,鉴定有希望的细菌物种进行同源cascade测试,然后从那些同源的crispr-cas系统获得或设计编码cas8和cascade的其他蛋白组分的多核苷酸序列,来鉴定另外的i-e型系统。
[0186]
编码来自许多物种(一些具有与来源于大肠杆菌菌株k-12mg1655的那些同源的cascade复合体)(列出在表3和表4中)的cascade的亚基蛋白的基因的多核苷酸dna序列,以及所得蛋白质组分的氨基酸序列,以及示例性最小crispr阵列,呈现为seq id no:22至seq id no:213(表3)。
[0187]
表3
[0188]
编码来自12种物种的cascade蛋白的基因的多核苷酸序列
[0189]
[0190]
[0191]
[0192]
[0193]
[0194]
[0195]
[0196]
[0197]
[0198]
[0199][0200]
蛋白质的多核苷酸序列来源于宿主细菌的gdna,并且针对在大肠杆菌中表达特异性地进行了密码子优化,和/或针对在真核细胞(例如,人类细胞)中表达特异性地进行了密码子优化。编码相应的最小crispr阵列的多核苷酸dna序列是基于来源于12种物种的重复序列,并且可以用于产生用作向导rnas的成熟的crrna。在表4中,最小crispr阵列包含在示例性的“间隔区”序列两侧的两条重复序列(小写的,有下换线的),其代表crrna的引导部分。通过核酸内切酶cascade亚基加工rna产生了于向导序列两侧的在5
’
和3
’
端上均具有重复序列的crrna。
[0201][0202]
在另一个实施方案中,本发明涉及编码来自具有其他i型表型的另外的细菌或古菌物种的cascade组分的改造的多核苷酸序列;包括但不限于i-b、i-c、i-f和i-f的变体类型,其可以根据本说明书的指导并通过使用生物信息学工具如blast和psi-blast来定位来自代表每种亚型的标志系统的cascade基因的同系物来进行鉴定和设计(参见,例如makarova,k.s.,et al.,nat.rev.microbiol.13:722-736(2015);koonin,e.v.,et al.,curr.opin.microbiol.37:67-78(2017))。在鉴定出所需的同系物之后,可以检查cascade
基因的两侧基因组邻域,以定位和鉴定本文公开的剩余cascade亚基蛋白的基因。例如,可以通过定位cas8同系物(并且可以通过定位cas5同系物来鉴定另外的i-f型变体2系统)和鉴定用于同源cascade测试的有希望的细菌物种,然后获得或设计编码来自那些同源的crispr-cas系统的cascade的cas8、cas5和其他蛋白组分的多核苷酸序列,来鉴定另外的i-f型系统。
[0203]
seq id no:214至seq id no:351示出了编码来自12种另外的同源cascade复合体的i-b、i-c、i-f和i-f变体第二类型的cascade的3种、4种或5种亚基蛋白的基因的多核苷酸dna序列,和所得蛋白组分的氨基酸序列,以及示例性的最小crispr阵列(表3)。亚基蛋白的多核苷酸序列来源于宿主细菌的gdna,并且针对在大肠杆菌中表达特异性地进行了密码子优化,和/或针对在真核细胞(例如,人类细胞)中表达特异性地进行了密码子优化。编码相应的最小crispr阵列的多核苷酸dna序列是基于来源于12种物种的重复序列,并且可以用于产生用作向导rnas的成熟的crrna。在表5中,最小crispr阵列包含在示例性的“间隔区”序列两侧的两条重复序列(小写的,有下划线的),其代表crrna的向导部分。通过核酸内切酶cascade亚基加工rna产生了于向导序列两侧的在5
’
和3
’
端上均具有重复序列的crrna。
[0204]
[0205][0206]
实施例19a至实施例19i和实施例22a至实施例22c描述了多种cascade复合体同系物的设计和测试,其每种包含cas亚基蛋白-foki融合蛋白,以评估每种cascade复合体的基因组编辑的效率。利用来自假单胞菌s-6-2的变体观察到最高的编辑,而其他同系物(即肠道沙门氏菌、地热杆菌epr-m、稻田甲烷胞菌mre50和嗜热链球菌(菌株nd07))则显示大约与大肠杆菌相等的编辑。利用改造的霍乱弧菌(vibrio cholera)菌株l15(i-f型)foki-cascade复合体和霍乱弧菌菌株he48(i-fv2型)foki-cascade复合体也观察到编辑。在一个实施方案中,这些不同同系物不同的pam要求能够增加靶标多核苷酸(例如,细胞中的gdna)中的靶标密度。因此,cascade复合体同系物的集合提供了靶标多核苷酸(例如,细胞中的gdna)中的核酸靶序列选择的更大的灵活性。
[0207]
在第二个方面,本发明涉及修饰的cascade亚基蛋白。适合修饰的cascade亚基蛋白包括但不限于本文所述物种的cascade亚基蛋白。
[0208]
在一个实施方案中,本发明涉及改造的循环排列的cascade亚基蛋白。此类循环排列的cascade亚基蛋白导致产生蛋白结构,其具有亚基蛋白的氨基酸的不同连通性的原始线性序列,但具有总体相似的三维形状(参见,例如bliven,s.,et al.,plos comput.biol.8:e1002445(2012))。循环排列的cascade亚基蛋白可以具有许多优点。例如,循环排列的cas7亚基蛋白可以产生新的n端和新的c端,其设计为被放置以用于与另外的多肽序列连接,以形成融合蛋白或连接子区域,而不破坏cas7蛋白折叠或cascade复合体组装。图3a和图3b中示出了循环排列的cas7(环状排列的cas7,cpcas7)的3个实例。在图3a和图3b中,显示了蛋白质的三个部分:天然蛋白质的n端部分(图3a,竖直条纹,例如,cas7蛋白)、天然蛋白质的中心部分(图3a,灰色阴影)和天然蛋白质的c端部分(图3a,无阴影)。图3a示出了将天然蛋白的n端部分重新定位到天然蛋白的c端位置,以产生环状排列的蛋白质(图3a,cpcas7),其中天然蛋白质的n端部分现在处于cpcas7的n末端,并通过连接子多肽连接到天然蛋白质的中央部分(图3a,连接子)。图3b示出了将天然蛋白质的c端部分(图3b,cas7)重新定位到天然蛋白质的n端位置(图3b,cpcas7),其中天然蛋白质的c端部分现在处于cpcas7的n末端,并且通过连接子多肽连接到天然蛋白质的中央部分(图3b,连接子)。
[0209]
实施例10a、实施例10b和实施例10中呈现的数据显示,包含循环排列的cas7亚基蛋白变体的cascade复合体的纯化证明了循环排列的i-e型crispr-cas亚基蛋白可以成功地用于形成具有与包含野生型蛋白的cascade复合体基本上相同的组合物(基于分子量)的cascade复合体。
[0210]
在另一个实施方案中,本发明涉及融合至另外的多肽序列以产生融合蛋白的cascade亚基蛋白,以及编码此类融合蛋白的多核苷酸。另外的多肽序列可以包括但不限于蛋白、蛋白域、蛋白片段和功能域。这样的另外的多肽序列的实例包括但不限于来源于转录活化剂或阻抑剂域和核苷酸脱氨酶(例如,胞苷脱氨酶或腺嘌呤脱氨酶,如komor,et.al.,nature553:420-424(2016);koblan,et.al.,nat.biotechnol.doi:10.1038/nbt.4172(may 29,2018)中所述的)的序列。本文中示出了融合蛋白另外的功能域。
[0211]
另外的多肽序列可以融合至任何cascade亚基蛋白中,其中所述另外的多肽序列由通常附接至包含cascade亚基蛋白的编码序列的多核苷酸的5
’
或3
’
端的另外的多核苷酸序列编码。在一些实施方案中,编码氨基酸连接子的另外的多核苷酸序列将cascade亚基蛋白连接至另外的目标多肽序列。在一些实施方案中,融合蛋白伴侣和连接子序列的多核苷酸序列可以来源于天然存在的gdna序列,或者可以针对在大肠杆菌中的细菌表达或在哺乳动物细胞(例如,人类细胞)中的真核表达进行密码子优化。在实施例1中示出了包含亲和标签(例如,his6,ii(iba gmbh llc,germany))、序列的核定位信号(nls)、麦芽糖结合蛋白和foki的融合蛋白的实例。实施例1中还公开了示例性的氨基酸连接子序列。
[0212]
实施例11a描述了cascade亚基蛋白-foki融合,以及cascade亚基蛋白与胞苷脱氨酶、核酸内切酶、限制酶、核酸酶/解旋酶或以上的域的融合。实施例11b描述了cascade亚基蛋白与其他cascade亚基蛋白的融合,以及cascade亚基蛋白与其他cascade亚基融合蛋白和酶促蛋白域的融合(实施例11d)。在一些实施方案中,可以在计算机中评估i型crispr亚
基蛋白用于在n端、c端或n端和c端之间的位置处产生蛋白融合的能力。在一些实施方案中,可以使用一个或多个多肽连接子,在n端、c端或n端和c端之间的位置处将i型crispr亚基蛋白连接至一个或多个融合域。在一些实施方案中,cascade亚基蛋白可以融合至单链foki(例如,单链foki与ecocascade rnp复合体融合;核苷酸序列,seq id no:1926;蛋白序列,seq id no:1927)。示例性的多肽连接子示出在实施例1、11、18和19中。
[0213]
图4a和图4b示出了包含融合至另外的蛋白序列(例如,foki)的cas8亚基蛋白(图4a,图4b,cas7、cas5、cas8、cse2、cas6,cas6周围的虚线框指示其与crrna发卡的相互作用;crna示出为包括发卡的黑线;以及cas8,指示了“c”c端,“n”n端)的cascade复合体。图4a显示了利用连接子多肽与cas8亚基蛋白的c端连接的(图4a,黑色的曲线)另外的蛋白序列(图4a,fp)的实例。图4b显示了利用连接子多肽与cas8亚基蛋白的n端连接的(图4b,黑色的曲线)另外的蛋白序列(图4b,fp)的实例。实施例11a描述了与foki核酸酶域n端融合的i-e型cas8的计算机设计、克隆、表达和纯化。
[0214]
图5a和图5b示出了包含融合至另外的蛋白序列的cascade亚基蛋白的cascade复合体的另外的实例。在图5a和图5b中,crna示出为包括发卡的黑线,并显示了cascade复合体的cas蛋白的相对位置(图5a,图5b:cas7、cas5、cas8、cse2、cas6;cas6周围的虚线框指示其与crrna发卡的相互作用)。图5a显示了每种经连接子多肽(图5a,黑色曲线)融合至6种cas7亚基蛋白中的每种的可检测部分(例如,绿色荧光蛋白;图5a,gfp)的实例。这样的cascade复合体可以用于通过由于与cascade复合体相关的多个可检测部分的存在而提供显著的信号扩增,来检测复合体与核酸靶序列的结合。图5b显示了利用连接子多肽与cas6亚基蛋白连接的(图5b,黑色曲线)另外的蛋白序列(图5b,fp)的实例。
[0215]
含有大肠杆菌i-e型cascade亚基蛋白的融合蛋白的实例包括但不限于以下:相同的亚基(例如,cse2_连接子_cse2)、环状排列的亚基(例如,cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7)、融合至核酸酶的i-e型cascade蛋白(例如,foki_连接子_cas8、cas3_连接子_cas8、cas6_连接子_foki、s1核酸酶_连接子_cse2_连接子_cse2)、融合至胞苷脱氨酶的i-e型cascade蛋白(例如,cas8_连接子_aid、cse2_连接子_cse2_连接子_apobec3g),以及i-e型cascade蛋白融合的一种或多种其他的i-e型cascade蛋白(例如,cas6_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7、cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cas5、cas6_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cpcas7_连接子_cas5)。
[0216]
图6a、图6b和图6c提供了包含cpcas7的改造的i型crispr-cas效应子复合体的图示。在图6a、图6b和图6c中,“cpcas7”是环状排列的cas7蛋白(图6a,图6b,图6c:cpcas7、cas5、cas8、cse2和cas6;cas6周围的虚线框指示其与crrna发卡的相互作用;crna示出为包括发卡的黑线;对于cpcas7,阴影对应于图3a中示出的环状排列的蛋白),并且显示了cascade复合体的cas蛋白的相对位置。图6a示出了包含6种单独的cpcas7亚基蛋白的cascade复合体(图6a,cpcas7)。图6b示出了包含6种融合的cpcas7亚基蛋白的cascade复合体,其中cpcas7亚基蛋白的c端(图6b,cpcas7)利用连接子多肽与邻近的cpcas7亚基蛋白的n端连接(图6b,连接子多肽示出为连接cpcas7亚基蛋白的深黑色线)。图6c示出了实施方案,其中cascade复合体包含6种融合的cpcas7亚基蛋白(“骨架”),其中第一cpcas7亚基蛋
159:647-61(2014))或阻抑剂域(例如,krab域)。在一些实施方案中,连接子核酸序列被用于连接蛋白、蛋白域或蛋白片段的两条或更多条编码序列。
[0222]
包含融合至转录活化剂的i型crispr-cas亚基蛋白的cascade复合体可用于激活基因的表达。靶标位点可包含转录起始位点(tss),该位点通常具有细胞的转录激活机制(因子)的一个或多个结合位点。图8示出了包含六种融合蛋白的cascade复合体,所述融合蛋白包含经连接子多肽(图8,将cpcas7与vp64连接的黑色曲线)连接到转录活化剂vp64的cpcas7(与图3a相比)。在图8中,crrna示出为包括发卡的深黑色线,并且显示了cascade复合体的cas蛋白的相对位置(图8:cpcas7、cas5、cas8、cse2和cas6;cas6周围的虚线框指示其与crrna发卡的相互作用)。这种cascade复合体的工程设计将复合体转换为用于基因的转录激活的灵活工具(cascadea),其中通过选择指导cascade复合体与选定基因的一个或多个调控元件(例如,tss)的结合的向导序列,实现了靶向选定的基因。实施例12描述了融合至vp64激活域以赋予cascade复合体转录激活活性的大肠杆菌i-e型cp-cas7蛋白的设计。转录活化剂包括但不限于:同源域蛋白、锌指蛋白、翼型螺旋(叉头)蛋白、亮氨酸拉链蛋白、螺旋环螺旋蛋白、异型二聚体转录因子、激活域,以及结合增强剂的转录因子(参见,例如molecular cell biology,harvey lodish,et al.,w h freeman&co;(2002)isbn 978-0849394805)。
[0223]
另外,包含融合至转录阻抑剂的i型crispr-cas亚基蛋白的cascade复合体可以用于抑制基因的表达。靶标位点可以包含转录调控元件。在一个实施方案中,cascade亚基蛋白可以经连接子多肽连接至krab域。包含cascade亚基蛋白/krab域融合的cascade复合体可以将复合体转化为用于基因转录抑制的灵活的工具(cascadei),其中通过选择指导cascade复合体与选定基因的一个或多个调控元件的结合的向导序列实现了靶向选定的基因。转录阻抑剂包括但不限于:被动转录阻抑剂、bzip转录因子家族、sp1样转录阻抑剂、活性转录阻抑剂(例如,经组蛋白脱乙酰基酶、组蛋白脱乙酰作用和双特异性阻抑剂的募集的转录抑制(参见,例如thiel,g.,et al.,eur.j.biochem.271:2855
–
2862(2004);nicola reynolds,n.,et al.,development 140:505-512(2013);gaston,k.,et al.,cell mol.life sci.,60:721-741(2003))。
[0224]
在另外的实施方案中,可以将cascade亚基蛋白融合至亲和标签。
[0225]
在本发明的其他实施方案中,可以通过插入选定的多核苷酸元件或向导多核苷酸内选定位置胡的核苷酸变化(例如,dna部分变化为rna部分的根本不同的变化,以及上述的向导多核苷酸的其他变化),来修饰i型crispr-cas向导多核苷酸。此类实施方案包括但不限于i型crispr-cas向导多核苷酸5'、3',或内部融合至一个或多个核苷酸效应子域(例如,ms2或ms2-p65-hsf1结合rna或募集转录因子的适体)。图9示出了i型crispr向导多核苷酸,并显示了cascade复合体的cas蛋白的相对位置(图9:cas7、cas5、cas8、cse2和cas6;cas6周围的虚线框指示其与crrna发卡的交互作用;crna示出为包括虚线框内的发卡的黑线)。在图9中,crrna还包含引入到向导多核苷酸的3'发卡中的rna适体发卡(图9,通过箭头指示的位置)。
[0226]
也可以修改i型crispr-cas向导的长度,通常通过延长或缩短cas7亚基蛋白和cse2亚基蛋白结合区域。图10a示出了具有3个cas7亚基、1个cse2亚基和缩短的crrna的cascade复合体(图10a:cas7、cas5、cas8、cse2和cas6;cas6周围的虚线框指示其与crrna发
卡的相互作用;crna示出为包括发卡的黑线)。图10b示出了具有9个cas7亚基、3个cse2亚基和延长的crrna的cascade复合体(图10b:cas7、cas5、cas8、cse2和cas6;cas6周围的虚线框指示其与crrna发卡的相互作用;crna示出为包括发卡的黑线)。
[0227]
实施例16描述了i型crispr-cas向导crrnas修饰的产生和测试,以及修饰的向导用于构建改造的i型crispr-cas效应子复合体的适合性。
[0228]
在第三个方面,本发明涉及编码一种或多种改造的cascade组分的核酸序列,以及包含编码一种或多种改造的cascade组分的核酸序列的表达盒、载体和重组细胞。本发明的第三个方面的一些实施方案包括的一种或多种多肽(例如,cse2、cas5、cas6、cas7和cas8蛋白,以及一种或多种同源向导),其中组分能够形成效应子复合体。通常,当表达一种以上的同源向导时,这些向导具有不同的间隔区序列以指导与不同的核酸靶序列的结合。这样的实施方案包括但不限于表达盒、载体和重组细胞。
[0229]
在一个实施方案中,本发明涉及一种或多种表达盒,其包含编码一种或多种改造的cascade组分的一种或多种核酸序列。表达盒通常包含涉及以下一项或多项的调控序列:转录调控、转录后调控或翻译调控。可以将表达盒引入多种生物体中,包括但不限于细菌细胞、酵母细胞、植物细胞和哺乳动物细胞(包括人类细胞)。表达盒通常包含对应于其所引入的生物体的功能调控序列。
[0230]
本发明的其他实施方案涉及包含编码一种或多种改造的cascade组分的一种或多种核酸序列的载体,包括表达载体。载体还可以包括编码可选择的或可筛选的标记的序列。此外,还可以将核靶向序列添加到例如cascade亚基蛋白中。载体还可以包括编码蛋白标签(例如,多聚his标签、血凝素标签、荧光蛋白标签和生物发光标签)的多核苷酸。可以将此类蛋白质标签的编码序列与例如编码cascade亚基蛋白的一种或多种核酸序列融合。
[0231]
用于构建表达载体的一般方法是本领域已知的。用于宿主细胞的表达载体是可商购获得的。有数种商业软件产品旨在促进适当载体的选择和其构建,如用于昆虫细胞转化和昆虫细胞中基因表达的昆虫细胞载体、用于细菌转化和细菌细胞中基因表达的细菌质粒、用于酵母和其他真菌中细胞转化和基因表达的酵母质粒、用于哺乳动物细胞转化和哺乳动物细胞或哺乳动物中基因表达的哺乳动物载体,以及用于细胞转化和基因表达的病毒载体(包括但不限于慢病毒、逆转录病毒、腺病毒、i型或ii型单纯疱疹病毒、细小病毒、网状内皮增生病毒和腺相关病毒(aav)载体),以用于细胞转化和基因表达以及易于允许克隆此类多核苷酸的方法。
[0232]
基于aav的载体(raav)是可用于本发明的方法实施的病毒载体的一个实例。aav是细小病毒科的单链dna成员,并且是一种天然的复制缺陷型病毒。aav载体是最常用于基因治疗的病毒载体。已知十二种人类aav血清型(aav血清型1[aav-1]至aav-12)和来自非人类的超过100种血清型。
[0233]
慢病毒载体是可用于本发明方法实施的病毒载体的另一个实例。慢病毒是逆转录病毒科的成员,并且是一种单链rna病毒,其既可以感染分裂细胞也可以感染非分裂细胞,并且可以通过整合到基因组中来提供稳定的表达。为了增加慢病毒载体的安全性,将产生病毒载体必需的组分分到多个质粒中。转移载体通常没有复制能力,并且可能在3
’
ltr中另外包含缺失,这使得整合后病毒会自行失活。包装和包膜质粒通常与转移载体结合使用。例如,包装质粒可以编码gag、pol、rev和tat基因的组合。转移质粒可包含病毒ltrs和psi包装
信号。包膜质粒通常包含包膜蛋白(通常是水泡性口炎病毒糖蛋白,vsv-gp,因为其感染范围广)。
[0234]
示例性的植物转化载体包括来源于根癌农杆菌(agrobacterium tumefaciens)的ti质粒的那些(参见,lee,l.y.,et al.,plant physiology146:325-332(2008))。同样,在本领域中有用和已知的是发根农杆菌(agrobacterium rhizogenes)质粒。例如,snapgenetm(gsl biotech llc,chicago,il;snapgene.com/resources/plasmid_files/your_time_is_valuable/)提供了载体、单个载体序列和载体图以及许多此类载体的商业来源的广泛的清单。
[0235]
为了在细菌表达系统中表达和纯化重组cascade,可以设计编码cascade亚基蛋白的载体,以及包含目标向导序列的最小crispr阵列。因此,本发明的一个方面包括这样的表达系统。在一个实施方案中,cascade复合体由三种不同的质粒载体表达,它们共同编码以下组分:cas8蛋白;cse2、cas7、cas5和cas6蛋白;以及crisprrna。在一些实施方案中,编码cas8的表达质粒包含天然的gdna基因序列,并且在其他实施方案中,表达质粒可以编码密码子优化的cas8以用于在选择的细胞类型中表达。类似地,编码cse2、cas7、cas5和cas6的表达质粒可以含有天然的gdna基因序列,或者可以含有已经进行密码子优化以用于在选择的细胞类型中表达的基因序列。在一些实施方案中,整个cascade亚基蛋白编码操纵子可以位于单个转录启动子下游,使得不同的蛋白质均从单个多顺反子转录物翻译而来。在另外的实施方案中,编码cascade亚基蛋白的基因可以彼此分隔开,具有介于中间的转录终止子和启动子。
[0236]
编码crrna的表达质粒可以在单个间隔区序列两侧、适当的转录启动子下游含有尽可能少的两个重复,或者可以在多个间隔区序列两侧含有同一个向导序列或多个不同的向导序列的许多重复。crispr和cascade亚基,特别是cas6亚基的协同表达导致将较长的前体crrna加工成成熟长度的crrna,其每个都包含在crrna的5'和3'端上的单个重复的片段,以及中间的单个间隔区序列。
[0237]
在大肠杆菌中表达完整cascade复合体的替代策略使用了两种质粒:一种质粒在单个表达质粒上编码整个cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子,并且一种质粒编码crisprrna。在这种情况下,通常与cas8基因的3'端重叠的cse2基因的5'端与cas8基因的3'端在空间上分开,以便附接编码亲和标签和/或蛋白酶识别序列的多核苷酸序列。
[0238]
实施例2描述了cascade蛋白的两种类型的细菌表达质粒系统:第一种类型包含两种质粒,第一种质粒编码cas8蛋白且第二种编码casbcde复合体的4种亚基蛋白(cse2
–
cas7
–
cas5
–
cas6操纵子);并且第二种类型包含编码cascade复合体的所有5种亚基蛋白(cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子)的表达质粒。还描述了同源crispr阵列。
[0239]
为了促进cascade复合体的纯化,可以将亲和标签附接到cse2亚基上,如n端strep-ii标签或六组氨酸(his6)标签。此外,可以将诸如tev蛋白酶或hrv3c蛋白酶之类的蛋白酶识别的氨基酸序列插入亲和标签和cse2亚基的天然n端之间,从而在最初纯化后,用蛋白酶对序列进行生物化学切割而从最终的重组cascade复合体释放出亲和标签。亲和标签也可以放置在其他亚基上,或留在cse2亚基上,并与其他亚基上的另外的亲和标签组合。包含亲和标签的示例性的cascade亚基蛋白示出在实施例1、实施例2、实施例3a、实施例3b和实施例3c中。
[0240]
对于i-e型cascade系统,可以利用编码crisprrna的质粒以及cse2
–
cas7
–
cas5
–
cas6基因转化大肠杆菌菌株,诱导蛋白表达,并可以产生缺少cas8亚基的cascade复合体。该cascade复合体通常被称为cas8-负cascade复合体,或者可选地称为casbcde复合体(参见,例如jore,m.,et al.,nat.struct.mol.biol.18:529-536(2011))。该纯化的复合体可以与单独纯化的cas8生物化学组合,以重构完整的cascade(参见,例如sashital,d.g.,et al.,mol.cell 46:606-615(2012))。
[0241]
表6示出了编码最小crispr阵列的细菌表达质粒的示例性的序列,cas8、cse2
–
cas7
–
cas5
–
cas6构建体,以及cas8
–
cse2
–
cas7
–
cas5
–
cas6构建体,其含有不同的标签和设计。可以遵循本说明书的指导,与在大肠杆菌k-12mg1655中发现的i-e型的示例性的表达质粒序列类似地设计编码cascade复合体的质粒和来自同源的i型系统的cascade复合体。表6另外含有表达cas8
–
cse2
–
cas7
–
cas5
–
cas6蛋白的表达质粒的序列,以及与cas8基因或cas6基因的foki融合,以用于产生用于基因编辑实验的核酸酶
–
cascade融合。
[0242][0243]
表7含有编码所有5种亚基蛋白的单一多聚启动子细菌表达质粒以及来自单个细菌表达质粒的crrna的序列。在该设计中,每个基因与其两侧的上游和下游具有转录启动子和终止子的其他基因分隔开。可以引入编码亲和标签和/或蛋白酶识别标签的另外的序列,以及与核酸酶蛋白的融合,以便产生cascade
–
核酸酶融合以用于基因编辑。
[0244][0245]
可以基于本文的设计标准,可以设计编码来自其他i型亚型和其他细菌或古菌生物体的同源的cascade复合体的另外的细菌表达质粒。可以利用cascade基因的gdna序列设计此类表达质粒,也可以利用已经进行密码子优化以用于在大肠杆菌或其他细菌菌株中表达的基因序列来设计它们。
[0246]
为了在诸如人类细胞的哺乳动物细胞中表达cascade或融合至cascade效应子,设计了真核表达质粒载体以使得能够通过真核转录和翻译机制表达相关的蛋白质和rna组分。在一个实施方案中,可以通过在真核启动子(例如,巨细胞病毒(cmv)启动子)驱动的单独的表达载体上编码每种蛋白质组分,并在rna聚合酶iii启动子(例如,人u6启动子)驱动的单独的表达载体上编码crrna,从而在哺乳动物细胞中产生cascade。crisprrna可以用最小crispr阵列编码,该阵列含有在一个或多个间隔区序列两侧的至少两个重复序列,这些序列用作成熟crrna的向导部分。可以设计产生crisprrna的构建体,其在最小阵列中的最外面重复序列两侧具有另外的序列。通过cascade复合体(cas6亚基蛋白)的rna处理亚基实现对crisprrna前体的处理,该rna可从单独的质粒表达。通过可以从单独的质粒表达的cascade复合体的rna加工亚基(cas6亚基蛋白)使得能够加工前体crisprrna。
[0247]
表8含有大肠杆菌i-e型cascade复合体的每种蛋白的单个真核表达质粒的序列。cas8亚基可以融合至另外的效应子核酸酶域,如foki核酸酶(实施例1,实施例3a,实施例3b和实施例3c)。表8还含有cascade的crrna组分的表达质粒的序列,其编码两种单独的crrnas,从而3个重复序列位于两个间隔间隔区两侧。可以将每个蛋白编码基因附接至附接核定位信号(nls)、亲和标签和连接那些标签的连接子序列的多核苷酸序列。可以通过通常附接至5
’
或3
’
编码序列的另外的多核苷酸序列,包括编码连接cascade亚基蛋白与另外的目标多肽序列的氨基酸连接子的另外的多核苷酸序列,来编码其他与任何cascade亚基蛋白的融合。本文中描述了候选融合蛋白的实例。
[0248][0249][0250]
为了在更少的表达载体上表达cascade复合体的组分,可以构建多顺反子表达载体,从而单个启动子(例如,cmv启动子)同时驱动多个编码序列的表达,这些编码序列被明脉扁刺蛾(thata asigna)病毒2a序列隔开。序列。2a病毒肽序列诱导核糖体跳跃,从而使多个蛋白质编码基因可在单个多顺反子构建体中串联起来,以在真核细胞中表达。因此,可以设计多顺反子载体,其在由单个启动子驱动的单个转录物上编码cascade复合体的4个或5个蛋白亚基。表9含有真核多顺反子表达质粒的序列,该序列可与crisprrna表达质粒组合以在哺乳动物细胞中产生功能性的cascade。
[0251][0252]
在一些实施方案中,crisprrna被编码在蛋白编码基因的3'非翻译区(utr)中,其表达由rna聚合酶ii启动子(例如,cmv启动子)驱动以产生转录物。在这样的实施方案中,最小crispr阵列被设计为存在于诸如cas6、cas7或报告基因(例如,增强型绿色荧光蛋白,egfp)之类的蛋白编码基因的下游,并通过先前已证明可赋予上游转录物以稳定性的malat1三链体序列与蛋白编码序列隔开。最小crispr阵列由cascade的rna加工亚基(通常使用不同的质粒表达)——一种切割最小crispr阵列的核酸内切酶加工,在转录物中引入断裂,并且三链体序列保护上游蛋白编码基因的3
’
端过早地被核酸外切降解。表10含有3个多核苷酸序列的序列,从而crispr序列被克隆到cas6、cas7或egfp的下游,并且整个融合序列的表达由cmv启动子驱动。
[0253][0254]
在一些实施方案中,crisprrna阵列被编码在与五个5cascade亚基蛋白表达的多顺反子构建体相同的载体上;这两种元件的组合产生了一个多合一的载体,该载体可产生cascade复合体的所有功能性亚基(蛋白质和rna),以及融合至一个cascade亚基的任何核酸酶或效应子域。表11含有这些多合一多核苷酸序列的两个代表性序列,其编码所有各自的组分以在哺乳动物细胞中产生功能性foki-cascade rnps。
[0255][0256]
实施例3a、实施例3b和实施例3c描述了使用表达每种cascade亚基蛋白的单独的质粒和最小crispr阵列的表达系统、其中从单个启动子表达多个cascade亚基蛋白编码序列的表达系统,以及其中构建了单个质粒cascade表达系统以表达用于哺乳动物细胞中的整个cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子和最小crispr阵列的表达系统。
[0257]
遵循本说明书的指导,本领域的普通技术人员可以设计编码类似于大肠杆菌i-e型cascade复合体中提供的实例的其他的cascade复合体的另外的哺乳动物表达载体。
[0258]
在第四个方面,本发明涉及通过将编码改造的i型crispr-cas效应子复合体的一种或多种组分的质粒引入宿主细胞中来产生改造的i型crispr-cas效应子复合体。转化的宿主细胞(或重组细胞)或已使用重组dna技术转染或转染的细胞后代可包含编码改造的i型crispr-cas效应子复合体的一种或多种组分的一种或多种核酸序列。将多核苷酸(例如,表达载体)引入宿主细胞中的方法是本领域已知的,并且通常根据宿主细胞的种类进行选择。这样的方法包括例如病毒或噬菌体感染、转染、结合、电穿孔、磷酸钙沉淀、聚乙烯亚胺介导的转染、deae-葡聚糖介导的转染、原生质体融合、脂质转染、脂质体介导的转染、粒子枪技术、微粒轰击、直接显微注射和纳米粒子介导的递送。在本发明的一个实施方案中,将编码改造的i型crispr-cas效应子复合体的组分的多核苷酸引入细菌细胞(例如,大肠杆菌)中。
[0259]
实施例4a和实施例4b描述了引入和表达cas8蛋白编码序列,以及改造的i型crispr-cas效应子复合体组分的编码序列的方法,以用于利用大肠杆菌表达系统经细菌产生此类复合体。
[0260]
本文公开的各种示例性的宿主细胞可用于使用改造的cascade效应子复合体产生重组细胞。这样的宿主细胞包括但不限于植物细胞、酵母细胞、细菌细胞、昆虫细胞、藻类细胞和哺乳动物细胞。
[0261]
为了便于讨论,下面使用“转染”指将多核苷酸引入宿主细胞中的任何方法。
[0262]
在一些实施方案中,利用编码i型crispr-cas效应子复合体的一种或多种组分的核酸序列瞬时或非瞬时转染宿主细胞。在一些实施方案中,像在对象中天然发生的那样转染细胞。在一些实施方案中,首先将被转染的细胞从对象,例如原代细胞或祖细胞中移除。在一些实施方案中,在离体转染后将原代细胞或祖细胞培养和/或返回到同一对象或不同的对象。
[0263]
改造的i型crispr-cas效应子复合体的表达和纯化是劳动密集型的,因此为了便于在大量的向导多核苷酸或效应子复合体变体上进行筛选,设计了更高通量的基于质粒的
递送系统。五个cas基因中的每一个都进行了人类密码子优化,并克隆到cmv驱动的表达质粒中,作为n端nls融合,并将含有靶向t细胞受体α位点(ucsc基因组浏览器,hg38)的traj27外显子的成对的grnas的最小crispr阵列克隆到人u6启动子下游的第六个质粒(实施例3a;图35)。在图35中,元件从左到右的顺序如下:hu6启动子、具菱形末端的灰色矩形;重复区1,空心菱形,(白色);间隔区1,灰色华夫格矩形;重复区2,灰色菱形;间隔区2,灰色点画矩形;以及重复区3,黑色菱形。在图35中,括号示出了编码两种grnas的区域。在一些实施方案中,两种向导rnas可以是相同的(例如,靶向相同的核酸靶序列),并且在其他实施方案中,两种向导rnas可以是不同的(例如,靶向两种不同的核酸靶序列)。
[0264]
大多数i型系统中的grna天然下是由cascade中出现的cas6核糖核酸酶催化的(参见,例如brouns,s.j.,et al.,science 321:960-964(2008);hochstrasser,m.,et al.,trends biochem.sci.40:58
–
66(2015),避免了对如本文所示的具有成对的grna的多个启动子方法的需要。因此,本发明的一个实施方案包括包含可操作地连接至调控元件以提供向导多核苷酸(例如,grnas)的表达的成对的向导多核苷酸的载体。在traj27位点处,六质粒共转染的产率高达~3%编辑,并且任一组分的去除废除了基因组编辑,唯一cas11除外,大肠杆菌cascade效应子复合体并非绝对需要cas11来进行dna结合(参见,例如westra,e.,et al.,rna biol.9:1134-1138(2012))。
[0265]
在本发明的另一实施方案中,最小crispr阵列,通常包含两种向导序列,并引入细胞或生化反应中作为dna模板。dna模板通过pcr扩增产生(例如,图42a;实施例20a)。这样的最小crispr阵列可以引入具有编码cascade复合体蛋白组分的一个或多个质粒的细胞中。在一些实施方案中,包含成对的向导多核苷酸的最小crispr阵列和载体均可以引入细胞或生化反应中。在使用两种cascade rnp复合体(例如,结合核酸靶序列的方法或切割核酸靶序列的方法;参见,例如图15a、图15b、图15c)的方法中,最小crispr阵列可以编码两种不同的向导。因此,在一些实施方案中,两种向导rnas可以是不同的(例如,靶向两种不同的核酸靶序列)。在使用单一cascade rnp复合体方法中(例如,当使用与mcas3蛋白相关的一种i型crispr-cas效应子复合体或其中cas3融合蛋白与复合体相关的i型crispr-cas效应子复合体时;例如,参见,例如图16a、图17b、图17c、图21a、图21b、图21c、图21d),最小crispr阵列可以编码相同向导序列的两个拷贝。因此,在一些实施方案中,两种向导rnas可以是相同的(例如,靶向相同的核酸靶序列)。
[0266]
在又一个实施方案中,可以将编码还包含被cas6蛋白识别以用于将crrna前体内切核苷酸加工为成熟的向导rnas的序列和结构的向导序列的多核苷酸引入细胞或生化反应中。在其他实施方案中,不需要加工的成熟的向导多核苷酸可用于级联复合体的组装。这种成熟的向导可以包含序列修饰(例如,在5'和/或3'端处的硫代磷酸酯键,以帮助保护向导免受核酸酶消化,如被rna酶消化)。另外的向导修饰包括本文针对核苷酸序列(例如,核苷酸类似物等)所述的那些修饰。
[0267]
实施例9a、实施例9b、实施例9c和实施例9d示出了包含foki融合蛋白的大肠杆菌i-e型cascade复合体的设计和递送,以促进人类细胞中的基因组编辑。实施例9b描述了递送表达cascade复合体组分的质粒载体至真核细胞中。在第五个方面,本发明涉及来自细胞的改造的i型crispr-cas效应子复合体的纯化,以及此类复合体的应用。改造的i型crispr-cas效应子复合体产生于宿主细胞中。从细胞裂解物纯化改造的i型crispr-cas效应子复合
体(在该情况下为cascade rnp复合体)。
[0268]
实施例5a和实施例5b描述了通过在细菌中过表达产生的大肠杆菌i-e型cascade rnp复合体的纯化,如实施例4b中所述的。该方法使用固定化金属亲和色谱,然后进行体积排阻色谱(sec)。实施例5a和实施例5b描述了可用于评估纯化的cascade rnp产品的质量的方法。提供的实例示出了cas8、cas7、cas6、cas5和cse2 cascade rnp复合体、包含cas7、cas6、cas5和cse2蛋白的cascade复合体以及foki-cas8融合蛋白的纯化。
[0269]
纯化的改造的i型crispr-cas效应子复合还可以直接用于生化测定(例如,结合和/或切割测定)。实施例6a、实施例6b和实施例6c描述了用于体外dna结合或切割测定的dsdna靶序列的产生。实施例6描述了产生靶序列的3种方法,包括合成的ssdna寡核苷酸的退火、选择的来自gdna的核酸靶序列的pcr扩增,以及克隆核酸靶序列到细菌质粒中。dsdna靶序列被用于cascade结合或切割测定中。
[0270]
在必要时,可以使用电泳迁移率变动测定(参见,例如garner,m.,et al.,nucleic acids res.9:3047-3060(1981);fried,m.,et al.,nucleic acids res.9:6505-6525(1981);fried,m.,electrophoresis 10:366-376(1989);fillebeen,c.,et al.,j.vis.exp.(94),e52230,doi:10.3791/52230(2014)),或实施例7中描述的生化切割测定,验证通过一种或多种改造的i型crispr-cas效应子复合体进行的位点特异性结合和/或切割。
[0271]
实施例7中所示的数据证明改造的i型crispr-cas效应子复合体可以显示近似定量的dna切割,如通过转化超螺旋、环形质粒底物为切割的线性形式所证实的。在利用改造的i型crispr-cas效应子复合体(例如,包含foki-cascade组分融合蛋白)证明强劲的生化活性后,在细胞中进行了基因组编辑。
[0272]
实施例8a、实施例8b、实施例8c和实施例8d示出了设计和递送包含cas亚基蛋白-foki融合蛋白的大肠杆菌i-e型cascade复合体至人类细胞。实施例8d中的数据证明了递送预组装的cascade rnps至靶标细胞中以及人类细胞中的有效基因组编辑。
[0273]
纯化的、改造后的i型crispr-cas效应子复合体可以直接引入细胞中。将组分引入细胞中的方法包括电穿孔、脂质转染、粒子枪技术和微粒轰击。
[0274]
图36a、图36b、图36c和图36d提供了使用改造的cascade-rnp复合体和改造的i型crispr-cas复合体的基于质粒的递送的人类细胞中的基因组编辑的比较性数据。在图36a-图36d,图36a中,用纯化的rnps转染hek293细胞,然后对编辑位点进行下一代测序(ngs)分析。如图36a(rnp转染)所示,将靶向两个相邻的位点foki-cascade rnp复合体(图36a,在图的左侧直线上方示出)核转染入hek293细胞中(图36a,图的左侧星形的、灰色的),以诱导dna切割和基因组编辑。计算了16个独特的基因组靶标位点处的编辑效率(见实施例6c、表31,人双hsa1-16)(n=1)。trac是t细胞受体的恒定区。当产生t细胞受体时,它们包括剪接连接(即“可变”区域和“连接”区域)。本文描述的一些trac向导靶向连接区域(例如,traj27)。每个靶标的间隔区间距显示在图下方(图36a,从左到右,25、30、35、40、45个碱基对(bp))。在图36a中,垂直轴为百分比编辑效率(图36a,编辑效率(%)),水平轴代表靶标1-16,且水平轴下方是以碱基对(bp)指示间隔区间长度的括号。
[0275]
图36b提供了图36a中靶标7的代表性dna修复结果。在图36b中,在图的顶部示出了成对的grnas靶向的半位点的相对位置及其相关的pam位点。间隔区间距通过顶行示出。在
图中,在顶部显示了预期的切割位点(图36b,位置“0”显示为垂直黑色中线)和bp距离(-50至50)。每条水平灰线代表在靶标位点观察到的不同类别的测序读长。这些线的指示符如下:灰色区域=序列匹配;水平黑线=缺失;且空心框=插入。位于图的每条线的右边都有一个圆圈:黑色圆圈是野生型读长;且白色空心圆圈是突变体读长。预期的野生型读长示出在第一灰色框(“ref”;即参考序列)中。野生型读长示出在第二灰色条(第二灰色条;图36b,黑色圆圈)中。接下来的11条线示出了突变体读长(图36b,空心圆)。以碱基对数量给出的插入长度显示在圆圈右侧的列中。读长的总百分比显示在右侧的下一栏中,且总读长示出在右侧的最后一栏中。
[0276]
如图36c中所示(6质粒转染系统),用6种质粒转染hek293细胞(图36c,星形的、灰色的,图的左侧),编码cas蛋白的5种质粒(图36c,指示为foki-cas8、cas11、cas7、cas5和cas6的质粒)和编码成对的grnas的一种质粒位于cmv和人u6(hu6)启动子控制之下(图36c,grna),然后进行编辑位点的ngs分析。foki-cascade rnp复合体的图示在虚线下方。计算了来自图36a的靶标7处的编辑效率(n=2)(图36a,图中黑条),并且包括缺乏单一组分的质粒混合物(图36c,水平轴以下,包含-/+的灰色框)作为对照(图36c,图中的空心条)。
[0277]
如图36d(2-质粒转染系统)中所示,用成对的grna表达质粒(图36d,grna质粒)和编码通过t2a“核糖体跳跃”序列肽分开的所有5种蛋白的多顺反子的表达质粒(图36d,cmv-cas7-2a-cas11-2a-cas5-2a-cas6-2a-foki-cas8)转染hek293细胞(图36d,星形的,灰色,图的左侧),然后进行编辑位点的ngs分析。foki-cascade rnp复合体的图示在虚线下方。针对来自图37c的2质粒系统转染(图36d,空心框)和的6质粒系统转染(n=3)(图36d,黑色条),计算了图36a中显示的16种靶标处的编辑效率。在图36d中,垂直轴是百分比编辑效率(“编辑效率(%)),水平轴代表靶标1-16,并且水平轴下方是以碱基(bp)对指示间隔区间长度的括号(图36d,从左至右,25、30、35、40、45bp)。
[0278]
实验是通过用在foki和cas6上含有核定位信号序列的纯化cascade-rnps对hek293细胞进行核转染来进行的。所通过对从gdna获得的pcr扩增子的下一代测序证明的,观察到高达~4%的编辑效率,并且在测试的16个靶标位点中,编辑通常位于包含30bp间隔区间长度的位点处(图36a)。对修复结果光谱的仔细检查揭示出,插入缺失聚集在间隔区的中间(图36b),与i型crispr-cas复合体的设计一致。因此,在本发明的一个实施方案中,将改造的i型crispr-cas复合体直接引入细胞中。对于6质粒递送实验(图36c),组装的质粒混合物包含420ng的除一种质粒外的每种质粒,然后在核转染后加水作为阴性对照或加入700ng缺失的质粒。对于最初的foki-ecocascade多顺反子的2质粒递送实验(图36d),将细胞分别用500ng的每种质粒或500ng成对的grna表达质粒和2.5μg多顺反子质粒进行电穿孔(每种条件总计3μg)。在一个实施方案中,所有五个cas基因被构建在通过t2a“核糖体跳跃”序列(参见,例如kim,j.,et al.,plos one 6,e18556(2011);liu,z.,et al.,sci.rep.7:2193(2017))串联连接的单个多顺反子的表达载体中(图36d)。令人惊讶的是,与多顺反子质粒和成对的grna表达质粒共转染产生的编辑效率和dna修复结果与利用6质粒方法(实施例9a)和直接rnp递送方法(实施例8a、实施例8b、实施例8c、实施例8d)观察到的那些类似,支持了以下结论,即生物化学地进行活性改造的i型crispr-cas效应子复合体被组装并运送到人类细胞的核中。总而言之,这些实验验证了大大简化的表达系统,其可以在真核细胞中利用大小与广泛使用的cas9和sgrna质粒相似的仅仅两种分子组分来重构精细的11亚基
rna引导的核酸酶。
[0279]
改造的i型crispr-cas复合体(大肠杆菌(ecocascade,假单胞菌s-6-2(psecascade)和铜绿假单胞菌(sthcascade))的数据表明,大多数靶标位点将是唯一的,因为它们必须包括两个半位点、必要的间隔区间距和允许的pam。选择了来自ecocascade、psecascade和sthcascade的改造的cascade同系物以进行更详细的定征。
[0280]
图37a、图37b、图37c和图37d示出了与foki连接子、间隔区间长度和cascade同系物相关的编辑效率。图37a,将foki-ecocascade编辑效率显示为foki-cas8连接子长度(图37a,空心圆圈,下面的线10aa;空心圆上图线,20aa;黑色的圆圈,17aa;和灰色的圆圈,30aa连接子长度)和间隔区间距的函数。在图37a中,垂直轴是编辑效率(%),且水平轴是以bp的间隔区间距。每个数据点代表平均3
–
4个独特的靶标位点。
[0281]
图37b提供了具有30-aa连接子的foki-cascade核酸酶。产生了12种i-e型cascade变体的foki-cas8连接子,并测试了4-7个靶标位点处的基因组编辑。每个数据点代表单个基因组位点,且条显示了位点间的均值和标准偏差(s.d.)。靶标含有aag(图37b,灰色条)或gaa(图37b,白色条)pam序列和30bp间隔区间距,其中水平轴上的物种如下:eco,大肠杆菌;pse,假单胞菌s-6-2;sen,肠道沙门氏菌;geo,地热杆菌epr-m;mar,稻田甲烷胞菌;ahe,阿兰蒂巴杆菌;oce,栖海洋菌hl-35;pae,铜绿假单胞菌;sth,嗜热链球菌;str,链霉菌s4;kpn,克雷伯氏肺炎菌;lba,毛螺菌科细菌。
[0282]
在图37c中,示出了foki-psecascade数据,其中垂直轴是百分比编辑效率(图37c,编辑效率(%)),且水平轴代表以碱基对(bp)的间隔区间长度。foki-cas8连接子长度为17个氨基酸。每个数据点代表单个基因组位点,并且条显示了7
–
8个位点间的均值和s.d.。
[0283]
图37d提供了foki-psecascade编辑效率作为pam序列的函数的数据,垂直轴是百分比编辑效率(图37d,编辑效率(%)),且水平轴对应于pam序列(图37d,从左至右,ccg、cgc、aag、aaa、atg、aac、agg、ata、gag和aat)。基因组位点含有一个aag pam,和在第二半位点处的可变的pam,如水平轴上所示的。每个数据点代表单个基因组位点,并且条显示了6
–
15个位点间的均值和s.d.。
[0284]
图37e提供了foki-ecocascade编辑效率(图37e,垂直轴,编辑效率(%))作为pam序列的函数的数据。靶标位点含有固定的aag pam,和在第二半位点处的可变的pam,如水平轴上所示的(图37e,从左至右,ccg、cgc、aag、agg、atg、gag、aaa、aac、ata和aat)。每个点代表hek293细胞中的单个靶标位点,且每个pam测试了6-15个位点(n=1/位点)。条形图显示了均值和s.d.。
[0285]
图37f提供了foki-sthcascade效率(图37f,垂直轴,编辑效率(%))作为pam序列的函数的数据。靶标位点含有固定的gaa pam,和在第二半位点处的可变的pam,如水平轴上所示的(图37f,从左至右,cc、aa、ga、ta和ca)。每个点代表hek293细胞中的单个靶标位点,且每个pam测试了18-33个位点(n=1/位点)。条形图显示了均值和s.d.。
[0286]
图37g提供的热图示出了来自图37c和图37d的显示较高的编辑效率(10
–
53%)的40个基因组位点的插入缺失类别频率。顶图的条形图中示出了0-60的百分比编辑效率。中间图中示出的热图显示了1
–
8bp的插入长度,且底图的热图中示出了1
–
50bp的缺失长度。在水平轴(1-40)上指示了40个基因组靶标位点(图37g,靶标)。单bp插入通过核苷酸同一性分离,且图底部的灰度强度标尺对应于插入频率百分比(图37g,ins freq(%),标尺为0至大
于或等于20)和缺失频率百分比(图37g,del freq(%),标尺为0至大于或等于20)。右边的条形图显示了每个插入缺失类别的平均频率(图37g,标尺为0到20)。右侧的饼图显示了从推定模板化修复产生的2
–
4bp插入的分数(图37g,饼图的黑色区域),此处定义为包含与切割位点相邻的重复序列。“其他”以饼图的灰色区域表示。
[0287]
研究了人类基因组中5个最高度编辑的foki-psecascade靶标位点(~20-48%编辑)的最密切相关的位点,仅受30-33bp的间隔区间要求的约束。在所有五个靶标中,在两个半位点中均未鉴定出<22错配的位点。对于foki-ecocascade foki-cas8连接子类型和间隔区间距实验(图37a),将细胞用2.4μg foki-ecocascade多顺反子质粒和~0.5-3.5μg成对的grna表达质粒进行核转染。
[0288]
对于foki-cascade同系物筛选(图37b),将细胞用1.5μg foki-cascade多顺反子质粒和~0.4-2.2μg成对的grna表达质粒进行核转染。在整个同系物中,靶向了4-7个位点,并选择了foki-ecocascade高编辑效率的位点。对于同系物变体foki-cas8连接子类型和间隔区间距编辑实验(图37c和图41a至图41c),将细胞用5μg多顺反子质粒和~100-400ng寡聚物模板化的成对的grna表达扩增子进行核转染。对于该实验,未对各孔或同系物变体中的grna浓度进行标准化。另外,对于图41a至图41c,将细胞用相比foki-ecocascade或foki-sthcascade grna平均~1.5x或更多的foki-psecascade grna进行核转染。
[0289]
本文描述了寡聚物模板化的pcr扩增(例如,实施例20a)。图42a和图42b中示出了在哺乳动物细胞中从人u6(hu6)启动子(图42a,420)产生用于成对的grna表达的扩增子的寡聚物模板化的pcr策略。简言之,反向内部寡核苷酸(图42a,424)编码两种grna序列,并针对新的靶标位点进行了修饰(也称为编码“重复区-间隔区-重复区-间隔区-重复区”序列的独特的引物(图42a,421:重复区,空心矩形;间隔区1,灰色矩形;重复区,空心矩形;间隔区2,灰色矩形;重复区,空心矩形),而剩余的引物是不变的(图42a:正向外部引物,422;正向内部引物,423;反向外部引物,425)。图42b中示出了在用编码foki ecocascade rnp复合体的多顺反子质粒和成对的grna表达质粒或成对的grna表达扩增子共转染hek293细胞后靶标7处的编辑效率(参见图36b)。在图42b中,垂直轴是编辑效率(%)且水平轴是成对的grna盒(ng)。数据点如下:分别为foki-ecocascade rnp复合体(ng)、成对的grna质粒、成对的grna扩增子;375,空心三角形,空心圆形;750,黑色三角形,黑色圆形;1,500,灰色三角形,灰色圆形;3,000,有白线的黑色三角形,有白线的黑色圆形。图42b中的数据证明了成对的grna表达扩增子相对于成对的grna表达质粒的相当的如果不是更高的编辑效率。
[0290]
对于pam筛选(图37d、图37e、图37f、图39a-图39d、图40c和图40f),通常,将细胞用3μg的foki-cascade多顺反子质粒和150ng(foki-psecascade和foki-ecocascade)或~80-120ng(foki-sthcascade)的寡聚物模板化的成对的grna表达扩增子进行核转染(除非另有说明)。
[0291]
为了进行特异性分析(图38a至图38c),将细胞用3μg多顺反子的cascade和150ng寡聚物模板化的成对grna表达扩增子进行核转染,并在核转染5天后收获。在图38a的顶部,水平线表示间隔区间距,剪刀表示预期的切割位点,且基因组靶标的半位点与其相应的pam区域一同显示(图38a,带有对比末端的矩形框)。通过虚线示出了所示半位点与靶标的关系。对于每个靶标,示出了32个碱基对,并且pam区域被示出为邻近种子序列。图38a提供了成对的grnas,其旨在包含与基因组靶标中一个或两个半位点错配的错配,如通过网格中的
填充框(不包括pam位点)所示的。注意,为简单起见,两个半位点都以相同的方向示出。图38b提供了针对错配的成对的grnas的每个组合的基因组靶标70处的相对编辑效率,其绘制为完美匹配的grnas的编辑效率百分比。在图38b中,顶行表示靶标(图38b,靶标70),下一行代表向导(图38b,grna1和grna2),下一行标识不匹配的集(图38b,mm集1和mm集2),下一行示出了foki-cascade rnp复合体。左栏示出了相对编辑向导1-mm集1/向导2-mm集2的数据,左栏示出了向导1-mm集2/向导2-mm集1的数据,两栏均示出了相对编辑效率百分比的数据(图38b相对编辑eff(%);标尺0-100),即左栏显示了grna1和grna2的数据,带有错配(mm)集1和2,且右栏显示了相同靶标的数据,但带有grna1和grna2之间的交换的错配(mm)集(n=1)。图38c提供了靶标73处的编辑效率(n=1),如图38b中所示的。
[0292]
在开发了通过寡聚物模板化的pcr扩增(如本文所述)产生成对的grna表达盒的、消除了对劳动密集型克隆步骤的需要的可扩展方法后,重新筛选了针对每种同系物变体的一组96个基因组靶标减的foki连接子和dna间隔区间长度。利用17-aa连接子,foki-psecascade在约30
–
33bp间隔区间窗内一致性地产生了平均~15-25%的编辑效率,并且一些靶标显示高达~40-50%的插入缺失(图37c)。利用其他同系物也观察到类似趋势。通过靶向包含一个同源pam和第二突变pam的基因组站点研究了pam要求。在体外已经表明,pam识别比刚性5
’-
gg-3
’
酿脓链球菌(s.pyogene)pam要求要混杂得多(参见,例如szczelkun,m.,et al.,proc.natl.acad.sci.usa 111:9798
–
9803(2014);hayes,r.,et al.,nature 530:499
–
503(2016);westra,e.,et al.,mol.cell.46:595
–
605(2012);fineran,p.,et al.,proc.natl.acad.sci.usa 111:e1629
–
e1638(2014);leenay,r.,et al.,mol.cell.62:137
–
147(2016))。令人惊讶的是,体外数据显示,确实有大量pams允许活性,出现了明显的等级次序偏向性(图37d;图39a至图39d)。相反,当突变的pam代表来自crispr阵列的“自我”靶标时,编辑被完全废除。
[0293]
在图39a-图39d的每个中,垂直轴对应于编辑效率(编辑效率(%)),且水平轴对应于与靶标相关的pam序列。图39a提供了作为pam序列函数的foki-psecascade编辑效率。基因组位点含有一个固定的atg pam,和在第二半位点处的可变的pam,如在水平轴上所示的。条显示了均值和s.d.(每个可变pam 6-14个位点,n=1/靶标位点)。注意,图37d描述了foki-psecascade的数据,其中一个pam固定在aag处,且另一个pam在包括atg在内的一组pams中是可变的。因此,那些pams的一个子集是aag-atg。图39a描述了foki-psecascade的数据,其中一个pam固定在atg处,且另一个pam在包括aag在内的pams集合中是可变的(图39a,水平轴,从左至右,aag、aac、aaa、atg、gag、ata、aat和agg)。因此,那些pams的一个子集还是aag-atg,并且是图37d中的相同aag-atg位点。
[0294]
图39b提供了作为pam序列的函数的foki-ecocascade编辑(图39b,水平轴,从左至右,ccg、cgc、aag、agg、atg、gag、aaa、aac、ata和aat)。固定的pam是aag,且条(bar)显示了均值和s.d.(每个可变pam6-15个位点,n=1/靶标位点)。图39c(图39c,水平轴,从左至右,aag、atg、aac、aaa、agg、gag、aat和ata)提供了与图39b所示相似的分析,但第一pam固定至atg(每个可变pam6-14个位点,n=1/靶标位点。图39b中的对应于aag-atg对(均值为~3)的atg栏与图39c中的对应于aag-atg对(均值也为~3)的aa栏相同。注意,垂直轴具有不同的标尺。图39d提供了作为pam序列的函数的foki-sthcascade编辑(图39d,水平轴,从左至右,cc、aa、ga、ta和ca)。固定的pam是gaa,且条显示了均值和s.d.(每个可变pam 18-33个位点;
n=1/靶标位点)。
[0295]
图40a、图40b、图40c、图40d、图40e和图40f示出了与改造的i型crispr-cas复合体的编辑效率的示例性的变化相关的数据。获得了图40a(foki-psecascade)和图40d(foki-sthcascade)中示出的百分比编辑效率(垂直轴)相对于以bps的间隔区间距(水平轴)的数据,基本上如实施例20c中针对图41a和图41c中所示的数据所述的。在图40a和图40d中,水平轴代表23-34bp间隔区间距,且图的条从左至右是17个aa(浅灰色条)、20个氨基酸(深灰色条)和30个aa(白色条)的foki-cas8多肽连接子长度。基本上按图39b所述的获得图40c和图40f中所示的数据。图40c和图40f提供了foki-psecascade和foki-sthcascade编辑(图40c,图40f,垂直轴,编辑效率(%))作为pam序列(图40c,从左至右,ccg、cgc、aag、aaa、atg、aac、agg、ata、gag和aat;图40f,从左至右,cc、aa、ga、ta和ca)的函数。图40b示出了foki-psecascade rnp复合体。foki-psecascade的固定的pam是aag(图40b,aag pam),且另一个pam在一组pams(图40b,可变的pam)中是可变的。图40e示出了foki-sthcascade rnp复合体。foki-sthcascade的固定的pam是gaa(图40b,gaa pam),且另一个pam在一组pams(图40e,可变的pam)中是可变的。重新筛选了foki-psecascade的连接子和间隔区间偏好性,且数据显示了近50%的编辑。还检查了pam偏好性。从该数据,确定了pams的体外等级次序偏好性。基本上,对嗜热链球菌的变体进行了相同的分析。在嗜热链球菌系统中,编辑较低。然而,本文所示的数据表明,在体内,在人类细胞中,嗜热链球菌系统对pam的偏好性非常混杂。前间隔区(即靶序列)上游的单个a允许进行编辑的事实,通常在基因中提供了增加数量的潜在靶序列(例如,相对于同一基因内的潜在的第二类ii型crispr-cas9 pam相关的靶标位点的数量)。此外,本文提出的体内数据与sinkunas,t.,et al.,embo j.32:385-394(2013)所证明的体外pam偏好性相关。
[0296]
在数百个经过编辑的基因组位点中累积的ngs数据提供了定征通过foki-psecascade引入的dsbs的dna修复结果的能力。集中于40个插入缺失频率>10%的独特位点,分析了缺失和插入的频率,作为围绕预测切割位点的50bp窗内总突变体读长的函数。2
–
4bp的插入高度富集,并且存在于检查的绝大多数位点中(图37e)。详细检查显示,这些插入中的~90%包含与切割部位相邻的序列的完美重复。尽管不希望受到任何特定理论的限制,但是这种重复可能是通过二聚体foki引入的交错切割的模板化修复的结果。
[0297]
通过利用大量错配的成对的grnas编辑两个高效率靶标位点,评估了foki-psecascade的特异性(图38a)。cascade的先前研究强调了~8-nt pam近端种子序列,以及32-nt向导grna中每第6个位置处的错配混杂,因为这些碱基被从在靶标结合后形成的rna-dna异源双链结构中翻转出来(参见,例如jung,c.,et al.,cell 170:35
–
47(2017);mulepati,s.,et al.,science 345:1479
–
1484(2014);fineran,p.,et al.,proc.natl.acad.sci.usa 111:e1629
–
e1638(2014);semenova,e.,et al.,proc.natl.acad.sci.usa 108:10098
–
10103(2011))。pam近端种子区域内的错配对基因组编辑高度有害,而pam远端的错配具有良好的耐受性,从而导致接近野生的编辑效率(图38b;图38c)。然而,当在两个半位点都出现了错配的区块时,在测试的整组成对的grnas中编辑急剧下降(图38b,图38c)。根据pam的数据和foki-psecascade介导的基因组编辑的和间隔区间数据(图38c;图37d),本发明的改造的i型crispr-cas复合体的一个优点是可靶向的位点能够在人类基因组中每~20至~30bp出现一次,而在潜在的脱靶位点处进行编辑是
不可能的。
[0298]
因此,在本发明的一个实施方案中,给定的改造的foki-cascade系统的潜在的可靶向位点或“靶标密度”是其有效间隔距离和pam偏好的函数,并且在同系物中将具有一些可变性。在一些实施方案中,可以使用以下标准来计算人类基因组中foki-psecascade、foki-ecocascade和foki-sthcascade的靶标密度(将数据外推以计算预测的靶标密度)。
[0299]
可以使用以下基序来计算foki-psecascade靶标密度:
[0300]5’–
[半位点1–
pam1]
–
[间隔区间]
–
[pam2–
半位点2]
–3’
。
[0301]
这里,[半位点1–
pam1]表示半位点1grna1靶标链靶序列和pam的反向互补物,并且[半位点2–
pam2]表示半位点2grna2非靶标链pam和靶标序列。基于支持以foki-psecascade进行编辑的间隔区间长度的分布(参见,例如图37d),有效的间隔区间长度为约30-33bp。pams被定义为属于给出最高编辑的集合1(aag、aaa、atg、aac),或者如果它们含有显示活性的任何测试pams,则属于集合2(aag、agg、atg、gag、aaa、aac、aat、ata)(参见,例如图39a;图40b)。据此,满足属于集合1或集合2的两种pams的优选的间隔区间长度的潜在靶标位点将分别平均每33.4bp或9.2bp出现一次。
[0302]
类似地测定foki-ecocascade的靶标密度,除了间隔区间长度定义为31-33个,并且pams定义为属于最高编辑的集合1(aag、agg、atg、gag、aaa),或者如果它们包含任何显示出活性的pams则属于集合2(aag、agg、atg、gag、aaa、aac、aat、ata)(参见,例如图39c;图39d)。据此,利用集合1pams或集合2pams计算了潜在的靶标位点,其分别平均每30.4bp或12.2bp出现一次。
[0303]
类似地测定foki-sthcascade的人基因组靶标密度,除了间隔区间长度定义为29-31bp,且pams定义为nna(参见,例如图39d)。据此,计算了潜在的靶标位点平均每4bp出现一次。
[0304]
因此,如本文所述,通过提供可用于基因组编辑的许多pam相邻靶序列,改造的i型crispr-cas复合体提供了一种提供各种潜在靶标位点的方法。因此,本发明的一个实施方案涉及使用与改造的i型crispr-cas复合体相关的pam序列在基因内提供增加数量的可用靶序列的方法(例如,相对于与第二类crispr-cas ii型或v型系统的pam序列相关的可用靶序列的数量)。该方法的应用涉及使用改造的i型crispr-cas复合体,其可以包括但不限于结合和/或切割靶序列、靶序列的突变、与靶序列或其调控元件相关的转录调控,以及通过使用本文所述的改造的i型crispr-cas复合体介导的靶序列,以及通过使用本文描述的改造的i型crispr-cas复合体介导的有意的修饰、变化和/或显著不同的结构变化(例如,在基因产物中)。
[0305]
在一些实施方案中,可以通过在基因组中的dna靶标位点处位点特异性地引入选择的多核苷酸序列(例如,一部分的供体多核苷酸)来产生gdna的修饰、改变和/或突变,将本文所述的改造的i型crispr-cas效应子复合体用于产生非人类的转基因生物体。转基因生物体可以是动物或植物。
[0306]
转基因动物通常是通过将改造的i型crispr-cas效应子复合体引入受精卵细胞中而产生的。参照转基因小鼠的制备描述的基本技术(参见,例如cho,a.,et al.,“generation of transgenic mice,”current protocols in cell biology,chapter.unit-19.11(2009))涉及五个基本步骤:首先,准备如本文所述的系统,其包括合
适的供体多核苷酸;第二,收获供体受精卵;第三,将系统显微注射到小鼠受精卵中;第四,将显微注射的受精卵植入假孕受体小鼠中;以及第五,进行基因分型并分析在首建小鼠中建立的gdna的修饰。首建小鼠会将遗传修饰传递给任何后代。首建小鼠通常是转基因的杂合子。这些小鼠之间的交配将产生在转基因25%的时间内为纯合子的小鼠。
[0307]
用于产生转基因植物的方法也是众所周知的,并且可以使用改造的1i型crispr-cas效应子复合体来应用。例如使用农杆菌介导的转化产生的转基因植物,通常含有一个插入一条染色体中的转基因。通过使含有单个转基因的独立的分离的转基因植物与其自己性交配(即自交),可能产生相对于转基因纯合的转基因植物。典型的接合性测定包括但不限于区分纯合子和杂合子的单核苷酸多态性测定和热扩增测定。
[0308]
在第六个方面,本发明涉及使用改造的i型crispr-cas效应子复合体来产生底物通道。在一些实施方案中,构建了包含底物通道元件和cas7亚基蛋白的融合蛋白。然后将这些cas7融合蛋白组装成改造的i型crispr-cas效应子复合体(例如,包含cse2、cas5、cas6、cas7底物通道元件融合和cas8)。在一些实施方案中,可以延伸改造的i型crispr-cas效应子复合体的crrna,以容纳另外的cas7亚基(参见,例如luo,m.,et al.,nucleic acids res.44:7385-7394(2016))。可以将不同的底物元件融合至cas7,然后以所需的化学计量混合。当这些各种cas7亚基组装成完整的i型crispr-cas效应子复合体时,底物元件的共定位可增强底物通道作用的功效。
[0309]
在一些实施方案中,构建了rna支架,使得在不存在其他i型crispr-cas效应子复合体组分的情况下,多个cas7底物通道元件融合体可以与其结合。
[0310]
底物通道元件可以融合至cas7的n端和/或cas7的c端。另外,可以将循环排列的cas7融合至底物通道元件。
[0311]
图11a和图11b示出了底物通道的图示,该通道由途径中的三种连续酶组成。底物通道有助于中间代谢产物直接传递至代谢途径链中连续酶的活性位点,而不会释放到额外的通道空间中。图11a示出了改造的底物通道的典型布置。酶e1、e2和e3与支架蛋白(s1、s2、s3)基质共价或非共价相互作用。双头箭头代表酶和支架蛋白之间的相互作用(例如亲和力相互作用)。然后将底物(x)加工成产品(y),而不会释放到额外的通道空间中。图11b示出了本发明的一个实施方案,其包括改造的i型crispr-cas效应子复合体,后者携带作为与cas7亚基蛋白的融合蛋白(即共价相互作用)的酶e1、e2和e3,从而产生底物通道。cpcas7蛋白和由cpcas7蛋白形成的骨架在本发明的该方面的实践中也可能是有用的。
[0312]
在其他实施方案中,底物通道元件可以融合至cas6。cascade复合体的cas6亚基可识别特定的rna发卡结构。可以构建由多个串联在一起的cas6 rna发卡结构组成的rna支架。来自不同cascade复合体的cas6肽具有不同的识别序列。因此,可以从多个正交的cas6 rna发卡构建rna支架。通过将不同的底物通道元件融合到正交cas6肽上,底物通道复合体可以以特定的化学计量组装。
[0313]
底物通道元件可以融合至cas6的n端和/或cas6的c端。另外,循环排列的cas6可以融合至底物通道元件。
[0314]
在一些实施方案中,目标异源代谢途径可以在模式生物,如大肠杆菌中表达。当基因被异源表达时,可以密码子优化基因以更有效地表达基因。
[0315]
在一个实施方案中,目标代谢途径是来自酿酒酵母的甲羟戊酸途径。该途径的底
物通道元件包括但不限于乙酰乙酰基-辅酶a-硫酶(atob)、羟基-甲基戊二酰-辅酶a合酶(hmgs)和羟基-甲基戊二酰-辅酶a还原酶(hmgr)。
[0316]
在另一个实施方案中,目标代谢途径是来自酿酒酵母的甘油合成途径。该途径的底物通道元件包括但不限于甘油3-磷酸脱氢酶(gpd1)和甘油-3-磷酸磷酸酶(gpp2)。
[0317]
在又一个实施方案中,目标代谢途径是来自梭状芽胞杆菌(clostridium stercorarium)的淀粉水解途径。该途径的底物通道元件包括但不限于cely和celz。
[0318]
在另外的实施方案中,目标代谢途径是来自大肠杆菌的葡萄糖磷酸转移酶途径。该途径的底物通道元件包括但不限于海藻糖-6-磷酸合成酶(tps)和海藻糖-6-磷酸磷酸酶(tpp)。
[0319]
在第七个方面,本发明涉及融合至cascade亚基蛋白的功能域被包含第二类ii型cas9蛋白和核酸靶向核酸(natna)的复合体的位点定向募集。本文公开了功能域,并且包括但不限于具有能够转录活化或能够转录抑制的酶促功能的蛋白域。实施例13a和实施例13b描述了以第一类i型crispr重复茎序列改造第二类ii型crisprsgrna、crrna、tracrrna或crrna和tracrrna序列,从而允许募集一种或多种cascade亚基蛋白至ii型crisprcas蛋白/向导rna复合体结合位点的方法。
[0320]
图12a、图12b和图12c示出了融合至cascade亚基蛋白的功能蛋白域被dcas9:natna复合体位点定向募集至靶标位点的一般化视图。包含间隔区序列(图12a,101)的第二类ii型crisprnatna(图12a,102)通过连接子核酸序列(图12a,103)共价连接至第一类i型crispr重复茎序列(图12a,104)。共价连接至i型crispr重复茎序列(图12a,105)的ii型crisrp natna能够结合至ii型dcas9(图12a,106)和i型cascade亚基蛋白(例如,cas6;图12a,107),其通过连接子序列(图12a,108)融合至功能蛋白域(例如,酶促域、转录活化或抑制域;图12a,109),从而形成rnp复合体。该rnp复合体(图12b,110)能够靶向包含与ii型crisprnatna间隔区序列(图12a,101)互补的靶序列(图12b,112)的双链dna(图12b,111)。rnp复合体的靶标识别导致间隔区序列(图12a,101)和靶序列(图12b,112)之间的杂交(图12b,113)。将cascade亚基功能域融合蛋白定位至dna允许通过临近基因的功能蛋白域或转录调控来修饰dna(图12c,114)。
[0321]
在第八个方面,本发明涉及包含改造的i型crispr-cas效应子复合体、改造的向导多核苷酸及以上的组合的组合物。在一些实施方案中,改造的i型crispr-cas效应子复合体包含相关的cas3融合蛋白。野生型i型crispr-cas系统需要用于dna靶向的cascade效应子复合体与用于进行性dna降解的cas3解旋酶-核酸酶的协同作用。在本发明的一个实施方案中,对i型crispr-cas效应子复合体进行改造以通过融合复合体至核酸酶域(例如,非特异性的foki核酸内切酶域)制备精确的dsbs。该方法使用靶向通过介于中间的序列(即间隔区间)分开的两个半位点dna序列的成对的向导多核苷酸。
[0322]
本发明的该方面的实施方案涉及包含两种改造的i型crispr-cas效应子复合体的组合物,所述复合体中的每种包含间隔区和包含cas亚基和核酸内切酶(例如,foki;参见,例如图2a、图2b和图2c的cascade复合体)的融合蛋白,其中至少两个参数是变化的以调节基因组编辑效率。这样的参数包括:
[0323]
用于产生包含cas亚基蛋白和核酸内切酶(例如,foki)的融合蛋白的连接子多肽的长度;以及
[0324]
间隔区能够结合的核酸靶序列之间的间隔区间距长度。
[0325]
本文提供了有关氨基酸组合物和序列连接子多肽的指导。
[0326]
本发明的该方面的一个实施方案是组合物,其包含:
[0327]
第一改造的i型crispr-cas效应子复合体,其包含:
[0328]
第一cse2亚基蛋白、第一cas5亚基蛋白、第一cas6亚基蛋白和第一cas7亚基蛋白,
[0329]
包含第一cas8亚基蛋白和第一foki的第一融合蛋白,其中第一cas8亚基蛋白的n端或第一cas8亚基蛋白的c端通过第一连接子多肽分别与第一foki的c端或n端共价连接,并且其中第一连接子多肽具有约10个氨基酸至约40个氨基酸的长度,和
[0330]
包含能够结合第一核酸靶序列的第一间隔区的第一向导多核苷酸;以及
[0331]
第二改造的i型crispr-cas效应子复合体,其包含:
[0332]
第二cse2亚基蛋白、第二cas5亚基蛋白、第二cas6亚基蛋白和第二cas7亚基蛋白,
[0333]
包含第二cas8亚基蛋白和第二foki的第二融合蛋白,其中第二cas8亚基蛋白的n端或第二cas8蛋白的c端通过第二连接子多肽分别与第二foki的c端或n端共价连接,并且其中第二连接子多肽具有约10个氨基酸至约40个氨基酸的长度,和
[0334]
包含能够结合第二核酸靶序列的第二间隔区的第二向导多核苷酸,其中第二核酸靶序列的前间区序列邻近基序(pam)和第一核酸靶序列的pam具有约20个碱基对至约42个碱基对的间隔区间距。
[0335]
这样的与第一核酸靶序列结合的第一改造的i型crispr-cas效应子复合体和与第二核酸靶序列结合的第二改造的i型crispr-cas效应子复合体的实例示出在图2a、图2b和图2c中。
[0336]
在一些实施方案中,第一连接子多肽和/或第二连接子多肽的长度为约15个氨基酸至约30个氨基酸,或者约17个氨基酸至约20个氨基酸的长度。在一个实施方案中,第一连接子多肽和第二连接子多肽的长度相同。
[0337]
第一cas8亚基蛋白和第二cas8亚基蛋白可以每个包含cas8亚基蛋白的相同的氨基酸序列。
[0338]
类似地,第一cse2亚基蛋白和第二cse2亚基蛋白可以每个包含cse2亚基蛋白的相同的氨基酸序列,第一cas5亚基蛋白和第二cas5亚基蛋白可以每个包含cas5亚基蛋白的相同的氨基酸序列,第一cas6亚基蛋白和第二cas6亚基蛋白可以每个包含cas6亚基蛋白的相同的氨基酸序列,第一cas7亚基蛋白和第二cas7亚基蛋白可以每个包含cas7亚基蛋白的相同的氨基酸序列,以及以上的组合。
[0339]
通常,第一cas8亚基蛋白的n端通过第一连接子多肽与第一foki的c端共价连接,第一cas8亚基蛋白的c端通过第一连接子多肽与第一foki的n端共价连接,第二cas8亚基蛋白的n端通过第二连接子多肽与第二foki的c端共价连接,第二cas8亚基蛋白的c端通过第二连接子多肽与第二foki的n端共价连接,以及以上的组合。
[0340]
本发明的该方面的实施方案包括实施方案,其中第二核酸靶序列和第一核酸靶序列之间的长度为约22个碱基对至约40个碱基对、约26个碱基对至约36个碱基对、约29个碱基对至约35个碱基对或约30个碱基对至约34个碱基对的间隔区间距。
[0341]
第一foki和第二foki可以是能够结合形成同型二聚体的单体亚基,或能够结合形成异型二聚体的不同的亚基。
[0342]
在优选的实施方案中,向导多核苷酸包含rna。
[0343]
在一些实施方案中,gdna包含第二核酸靶序列的pam和第一核酸靶序列的pam。
[0344]
在一些实施方案中,改造的i型crispr-cas效应子复合体基于选自以下的一种或多种生物体的i型crispr-cas效应子复合体:肠道沙门氏菌、地热杆菌(菌株epr-m)、稻田甲烷胞菌mre50、铜绿假单胞菌(例如,铜绿假单胞菌(菌株nd07)、假单胞菌s-6-2和大肠杆菌。在优选的实施方案中,改造的i型crispr-cas效应子复合体基于铜绿假单胞菌(例如,铜绿假单胞菌(菌株nd07)、假单胞菌s-6-2和/或大肠杆菌的i型crispr-cas效应子复合体。假单胞菌s-6-2相比大肠杆菌同系物诱导~10倍高的编辑效率,并且测试的其他同系物的大约一般显示与大肠杆菌同等水平的活性,证明了来自不同的i型系统的改造的i型crispr-cas效应子复合体可以在功能上用于人类细胞中的基因组编辑。
[0345]
实施例18a、实施例18b、实施例18c、实施例18d、实施例20a、实施例20b和实施例20c中所示的数据证明了改变用于产生包含cas亚基蛋白和foki的融合蛋白的连接子多肽的长度和/或改变间隔区能够结合的核酸靶序列之间的间隔区间距长度促进了细胞中基因组编辑效率的调节。
[0346]
在又一个实施方案中,本发明涉及改造的i型crispr-cas效应子复合体,其包含含有cascade亚基蛋白(例如,cas8亚基蛋白)和第一功能域(例如,foki)的第一融合蛋白,以及包含dcas3*蛋白和第二功能域(例如,foki)的第二融合蛋白(图13a:cas7、cas5、cas8、cse2和cas6,cas6周围的虚线框指示了其与crrna发卡的相互作用;crna示出为包括发卡的黑线)。包含第一功能域(例如,foki)(图13a,cas8-连接子1-fp1融合)的改造的i型crispr-cas效应子复合体可以结合dna,并且然后可以募集dcas3*-第二功能域(例如,foki)融合蛋白(图13a,dcas3*-连接子2-fp2)。在其中第一功能域(图13a,cas8-连接子1-fp1融合)和第二功能域(图13a,dcas3*-连接子2-fp2)包含二聚体蛋白的亚基的情况下,dcas3*-第二功能域(例如,foki)融合蛋白结合包含第一功能域(例如,foki)的改造的i型crispr-cas效应子复合体,促进第一功能域和第二功能域的二聚化(图13a)。图14a示出了与包含经连接子多肽(图14a,连接子1)连接至cas亚基蛋白(图14a,有条纹的框)的第一功能域(图14a,fd1)和经与cascade复合体相关的连接子多肽(图14a,连接子2)连接至第二功能域(图14a,fd2)的dcas3*的改造的i型crispr-cas效应子复合体(图14a,cascade)的dsdna的结合;从而使得fd1和fd2接近并促进fd1和fd2的相互作用。cascade复合体的结合涉及单个pam序列(图14a,pam,空心框)。在图14a中,dsdna示出为成对的水平虚线。在功能域为二聚体核酸内切酶(例如,foki)的情况下,fd1和fd2的接近有利于形成功能二聚体。
[0347]
本发明实施方案的一个优点是,相对于使用两种foki-cascade复合体(将图14a与图2a、图2b和图2c进行比较),可以使用单一cascade复合体(识别单一pam序列)来切割双链核酸靶序列。使用两种foki-cascade复合体需要适当定向的两个pam序列(图2a、图2b和图2c),这可能会限制对近端核酸靶序列的选择。
[0348]
用于产生包含cas亚基蛋白和核酸内切酶(例如,foki)的融合蛋白的连接子多肽的长度和/或组合物,以及用于产生包含dcas3*蛋白和核酸内切酶的融合蛋白的连接子多肽的长度和/或组合物可以改变,以调节基因组编辑效率。实施例21a、实施例21b、实施例21c和实施例21d描述了多种cas3-foki连接子组合物和长度的设计和测试,以及用于调节基因组编辑效率的foki-cas8连接子组合物和长度。
[0349]
本发明的该方面的另一个实施方案包括改造的i型crispr-cas效应子复合体(图13b:cas7、cas5、cas8、cse2和cas6;cas6周围的虚线框指示其与crrna发卡的相互作用;crna示出为包括发卡的黑线),以及包含通过连接子多肽(图13b,连接子)连接的dcas3*蛋白(图13b,dcas3*)和功能域(图13b,fp)(例如,胞苷脱氨酶)的融合蛋白。改造后的i型crispr-cas效应子复合体可以结合dna并募集dcas3*-功能域(例如,胞苷脱氨酶)融合蛋白。该实施方案可以促进核酸靶序列的位点特异性靶向,以用于通过功能域进行修饰或与功能域相互作用。在胞苷脱氨酶的情况下,可将改造的i型crispr-cas效应子复合体和包含dcas3*蛋白和胞苷脱氨酶的融合蛋白用于核酸靶序列中的位点特异性碱基编辑。图14b示出了改造的i型crispr-cas效应子复合体(图14b,cascade)的实例,其包含融合蛋白,该融合蛋白包含经连接子多肽(图14b,连接子)与功能域(图14b,fd)连接的dcas3*蛋白(图14b,dcas3*),其中复合体结合到dsdna(图14b,成对的水平虚线)。在图14b中,功能域与dsdna的接触得到促进。cascade复合体的结合涉及单个pam序列(图14b,pam,空心框)。图14c示出了改造的i型crispr-cas效应子复合体(图14c,cascade)的另一实例,其包含融合蛋白,所述融合蛋白包含经连接子多肽(图14c,连接子)与功能域(图14c,fd)连接的dcas3*蛋白(图14c,dcas3*),其中复合体结合至dsdna(图14c,成对的水平虚线)。cascade复合体的结合涉及单个pam序列(图14c,pam,空心框)。在图14c中,功能域与ssdna的接触得到促进。
[0350]
可用于构建与i型crispr-cas亚基蛋白的融合蛋白的另外的功能域和蛋白在本说明书和实施例中有描述。可以遵循实施例21a至实施例21d及本说明书的指导评估连接子多肽组合物和cas3-连接子多肽-功能域融合蛋白的长度,以评估对功能域性能的影响。
[0351]
本发明的一些实施方案可以使用改造的i型crispr-cas效应子复合体和mcas3蛋白,其中mcas3蛋白包含下调的解旋酶活性(例如,mcas3蛋白—一种cas3进行性突变体蛋白,相对于野生型i型crisprcas3蛋白具有沿dna的减少的移动),或者mcas3蛋白缺乏解旋酶活性(例如,mcas3蛋白不再是进行性的核酸酶如wtcas3蛋白,但mcas3蛋白保留了切口活性)。改造的i型crispr-cas效应子复合体可以结合dna然后募集mcas3蛋白。该实施方案可以促进基因组dna的位点特异性的切割。
[0352]
表48描述了大量的mcas3蛋白,其中对cas3蛋白进行的突变影响解旋酶域的atp结合/水解区域或者解旋酶域的ssdna通路保守区。图44显示了ecocas3蛋白的功能域的线性示图和在cas3编码序列中制作的突变的相对位置。在图44中,指示了hd核酸酶域(氨基酸1-272)、解旋酶域(reca1区域,氨基酸273-521;reca2区域,氨基酸522-737)、连接子(氨基酸738-794)和c端域(ctd,氨基酸795-888)。huo,y.,et.al.,nat.struct.mol.biol.9:771-777(2014)提供了利用来自褐色嗜热裂孢菌(thermobifida fusca)(登录码:q47pj0;seq id no:1869)、绿色糖单孢菌(saccharomonospora viridis)(c7mta6;seq id no:1870)、弯曲高温单孢菌(thermomonospora curvata)(d1a6q2;seq id no:1922)、阿佛曼链霉菌(streptomyces avermitilis)(q825b5;seq id no:1925)、波卓链霉菌(streptomyces bottropensis)(m3di13;seq id no:1923)、嗜热栖热菌(thermus thermophiles)菌株hd8(q53vy2;seq id no:1924)和大肠杆菌(p38036;seq id no:1844)的蛋白的cas3家族的序列比对进行的序列保守性分析。筛选了在解旋酶域或ssdna环结合域的atp结合部分中具有突变的24种不同的ecocas3蛋白变体(实施例23a至实施例23c)。7种突变体在扩增子窗中显示明显更多和/或位置变动的缺失类别;该发现支持了那些mcas3蛋白相对于wtcas3具有减
少的进行性。
[0353]
实施例23a至实施例23c描述了这样的mcas3蛋白,其中相对于利用相应的wtcas3蛋白产生的平均的缺失,平均的mcas3蛋白诱导的缺失更短。这样的mcas3蛋白可用于基因组编辑(例如,在人类细胞中)。图45a、图45b、图45c和图45d示出了指示mcas3蛋白的数据,当引入和在人类细胞中表达时,当与cascade rnp复合体相关时其相对于与cascade rnp复合体相关的wtcas3蛋白产生更短的平均缺失长度。参考本说明书的教导,本领域普通技术人员可以在从除大肠杆菌以外的其他细菌物种获得的cas3蛋白的相应区域中进行相似的突变。
[0354]
实施例26a至实施例26c提供了可用于产生基因组缺失的mcas3蛋白的另外的实例,其中平均mcas3蛋白诱导的缺失相对于利用相应的wtcas3蛋白产生的平均缺失更短。实施例中示出的数据支持来自假单胞菌s-6-2的cas3的atp酶/解旋酶缺陷型变体(mpsecas3蛋白)可以与psecascade rnp复合体一起使用,来在预期的切割位点处产生缺失(即切割位点定位的缺失)。
[0355]
进一步定征了wtpsecas3蛋白/psecascade活性。使用使得能够检测大基因组缺失的靶标富集探针进行了另外的实验。具体地,基本上按照实施例26a至实施例26c中所述,用编码psecascade rnp复合体、wtpsecas3蛋白和针对trac位点的最小crispr阵列的dna模板转染hek293细胞。靶标富集探针用于分离和测序基因组片段;而在实施例26c中,使用扩增子窗口来识别缺失的存在。靶标富集/测序方法提供了较大缺失的无偏见观察,其通过使用扩增子窗来识别缺失无法提供。总体而言,发现使用靶标富集和基因组片段测序评估的缺失在很大程度上是单向的,从wtpsecas3蛋白起始位点的上游开始。缺失范围从1bp到近250kb。除了提供切割基因组dna的方法并提供给定长度的缺失外,该方法还可用于在确定的位置生成缺失的较大的随机子集,以探测基因的调控/启动子区域。
[0356]
mcas3蛋白可以包含一个或多个突变(例如,如表48中所述的突变的组合)。
[0357]
证明了几种mcas3蛋白的缺失长度的控制。在一些实施方案中,本发明的与包含向导多核苷酸的cascade复合体相关的mcas3蛋白可以提供约1个至约600个碱基对、约1个至约500个碱基对、约1个至约400个碱基对、约1个至约300个碱基对,优选约1个至约250个碱基对、约1个至约200个碱基对或约1个至约100个碱基对的平均缺失长度。
[0358]
在一些实施方案中,wtcas3蛋白或mcas3蛋白可以融合至cascade复合体的各种亚基以进一步控制cas3平均缺失长度。约束cascade复合体可以限制或防止cas3蛋白或mcas3蛋白沿dna移动,因为其将被固定至cascade复合体结合的位点。通常可以利用连接子多肽将wtcas3蛋白或mcas3蛋白融合至cascade复合体的蛋白组分的n端或c端域(例如,对于ecocascade复合体,融合可以为与ecocas8、ecocas6或ecocas5)。nls序列也可以附接至融合蛋白的n端。表12中示出了大肠杆菌cascade蛋白组分的此类构建体的实例。这些ecocas3融合蛋白还具有附接至其n端的nls序列。
[0359][0360][0361]
*蛋白序列是编码的多顺反子的蛋白序列
[0362]
本发明的实施方案包括能够相对于野生型i型crisprcas3蛋白(wtcas3蛋白)减少沿dna的移动的改造的i型crisprmcas3蛋白。在一些实施方案中,mcas3蛋白包含与相应的wtcas3蛋白的约90%或更高的,优选约95%或更高的,更优选约98%或更高的序列同一性。mcas3蛋白的编码序列可以在氨基端、羧基端或氨基端和羧基端包含共价连接的核定位信号。mcas3蛋白可以包含下调解旋酶活性的一个或多个突变,其中改造的mcas3蛋白相对于相应的wtcas3蛋白保留了核酸酶活性(或其至少一部分)。通常,dna是包含含有核酸靶序列的靶区的dsdna。当wtcas3蛋白与相应的cascade核蛋白复合体(“cascade np复合体/wtcas3蛋白”;例如,cascade rnp复合体)相关,并且cascade np复合体包含含有与核酸靶序列互补的间隔区的向导时,cascade np复合体/wtcas3蛋白与核酸靶序列的结合有利于dna靶区中的切割,通常导致靶区中的缺失;并且mcas3蛋白当其与cascade np复合体(“cascade np复合体/mcas3蛋白”;例如,cascade rnp复合体/mcas3蛋白)相关并且结合核酸靶序列时,有利于dna靶区中的切割,并导致相对于wtcas3平均缺失长度更短的平均缺失长度。
[0363]
在一些实施方案中,mcas3蛋白中的一个或多个突变是相对于wtcas3蛋白的氨基酸取代。在其他实施方案中,一个或多个缺失包括mcas3蛋白编码序列中相对于wtcas3蛋白的氨基酸缺失或插入。一个或多个突变可以位于解旋酶域的reca1区域或reca2区域中。在一个实施方案中,相对于wtcas3蛋白(例如,影响ssdna环结合的突变和/或解旋酶域的ssdna通路保守区域中的突变),一个或多个突变下调mcas3蛋白与ssdna的结合。在另外的实施方案中,相对于wtcas3蛋白,一个或多个突变下调通过mcas3蛋白的atp水解,或者相对于wtcas3蛋白,下调atp与mcas3蛋白的结合。在其他实施方案中,mcas3蛋白包含一个或多个突变的组合,其相对于wtcas3蛋白下调mcas3蛋白与ssdna的结合、相对于wtcas3蛋白下调通过mcas3蛋白的atp水解,或下调atp与mcas3蛋白的结合。
[0364]
其他实施方案包括共价连接至cascade核蛋白复合体(例如,cascade rnp复合体)
的cas蛋白的编码序列的氨基端或羧基端的mcas3蛋白的编码序列。这样的cas蛋白可以选自cse2、cas8蛋白、cas7蛋白、cas6和cas5蛋白。
[0365]
在一些实施方案中,wtcas3蛋白是大肠杆菌1型crisprcas3蛋白。在其他实施方案中,wtcas3蛋白是选自假单胞菌s-6-2、褐色嗜热裂孢菌、绿色糖单孢菌、弯曲高温单孢、阿佛曼链霉菌、波卓链霉菌、嗜热栖热菌、霍乱弧菌、肠道沙门氏菌、地热杆菌epr-m、稻田甲烷胞菌mre50和铜绿假单胞菌(菌株nd07)的wtcas3蛋白。
[0366]
对于大肠杆菌1型crisprwtcas3蛋白,一个或多个突变可以包括但不限于d452h、a602v或d452h和a602v。
[0367]
在其他实施方案中,细胞包含dna,其中细胞可以是真核细胞(例如,人类细胞)。
[0368]
在另外的实施方案中,本发明包括包含mcas3蛋白的编码序列的多核苷酸、包含mcas3蛋白编码序列的表达盒、包含mcas3蛋白编码序列的质粒,以及包含mcas3蛋白的cascade核蛋白复合体。
[0369]
在第九个方面,本发明涉及使用改造的i型crispr-cas效应子复合体的方法。
[0370]
在一些实施方案中,本发明包括结合多核苷酸(例如,dsdna)中的核酸靶序列的方法,包括提供一种或多种改造的i型crispr-cas效应子复合体,以用于诱导至细胞或生化反应中,以及将改造的i型crispr-cas效应子复合体引入细胞或生化反应中,从而促进改造的i型crispr-cas效应子复合体与多核苷酸的接触。复合体与多核苷酸的接触导致改造的i型crispr-cas效应子复合体与多核苷酸中的核酸靶序列的结合。
[0371]
在一个实施方案中,改造的i型crispr-cas效应子复合体包含与多核苷酸中的核酸靶序列互补的向导。改造的i型crispr-cas效应子复合体结合多核苷酸中的核酸靶序列。
[0372]
在其他实施方案中,第一改造的i型crispr-cas效应子复合体包含与多核苷酸中的第一核酸靶序列互补的向导,并且第二改造的i型crispr-cas效应子复合体包含与多核苷酸中的第二核酸靶序列互补的向导。第一改造的1i型crispr-cas效应子复合体结合第一核酸靶序列,并且第二改造的i型crispr-cas效应子复合体结合多核苷酸中的第二核酸靶序列。
[0373]
在又一个实施方案中,改造的i型crispr-cas效应子复合体包含与多核苷酸中的核酸靶序列互补的向导,并且还包含能够与复合体结合的dcas3*融合蛋白。改造的i型crispr-cas效应子复合体结合多核苷酸中的核酸靶序列,并且效应子复合体包含与复合体结合的dcas3*融合蛋白。
[0374]
此类结合核酸靶序列的方法可以在体外进行(例如,在生化反应中或在培养的细胞中;在一些实施方案中,培养的细胞是仍保留在培养物中且不会引入人体内的人类培养细胞);体内进行(例如,在活生物体的细胞中,附带条件是,在一些实施方案中,该生物体是非人的生物体);或离体下进行(例如,从对象去除的细胞,附带条件是,在一些实施方案中,该对象包括人类对象,且在其他实施方案中,该对象是非人类对象)。
[0375]
本领域已知评估和/或定量核酸序列与核苷酸之间的相互作用的各种方法,包括但不限于以下各项:免疫沉淀(chip)测定、dna电泳迁移率变动测定(emsa)、dna下拉测定,以及微孔板捕获和检测测定。商业试剂盒、材料和试剂可用于实施许多这样的方法,并且例如,可以从以下供应商处获得:thermo scientific(wilmington,de)、signosis(santa clara,ca)、bio-rad(hercules,ca)和promega(madison,wi)。检测多肽和核酸序列之间的
相互作用的一种常用方法是emsa (参见,例如hellman l.m.,et al.,nature protocols 2:1849-1861(2007))。
[0376]
在另一实施方案中,本发明包括切割多核苷酸中的核酸靶序列(例如,dsdna中的单链切割或dsdna中的双链切割)的方法,包括提供用于用到至细胞或生化反应中的一种或多种改造的i型crispr-cas效应子复合体,以及将改造的i型crispr-cas效应子复合体引入细胞或生化反应中,从而促进改造的i型crispr-cas效应子复合体与多核苷酸的接触。
[0377]
在一个实施方案中,将包含与多核苷酸中的第一核酸靶序列互补的向导和第一核酸酶域(例如,foki)的第一改造的i型crispr-cas效应子复合体(图15a,cascade1,实线轮廓的框,经连接子多肽连接,黑色曲线,至第一核酸酶域,表示为圆扇形),以及包含与多核苷酸中的第二核酸靶序列互补的向导和第二核酸酶域(例如,foki)的第二改造的i型crispr-cas效应子复合体(图15a,cascade 2,虚线轮廓的框,经连接子多肽连接,黑色曲线,至第二核酸酶域,表示为圆扇形)引入细胞或生化反应中。第一改造的i型crispr-cas效应子复合体(图15b,cascade1)结合dsdna(图15b,通过成对的水平黑线表示的dsdna)中的第一核酸靶序列,并且第一核酸酶域切割dsdna的第一链(图15c,cascade1),并且第二改造的i型crispr-cas效应子复合体(图15b,cascade2)结合dsdna中的第二核酸靶序列,并且第二核酸酶域切割dsdna的第二链。改造的i型crispr-cas效应子复合体的结合导致多核苷酸(例如,dsdna)中的核酸靶序列被改造的i型crispr-cas效应子复合体切割。
[0378]
在另外的实施方案中,将第一改造的i型crispr-cas效应子复合体包含与多核苷酸中的第一核酸靶序列互补的向导,包含与多核苷酸中的第二核酸靶序列互补的向导的第二改造的i型crispr-cas效应子复合体,以及cas3切口酶(例如,仅具有切口酶活性的atp酶缺陷型cas3变体)引入细胞或生化反应中。第一改造的i型crispr-cas效应子复合体结合dsdna中的第一核酸靶序列,cas3切口酶蛋白与第一复合体相关,并切割dsdna的第一链,并且第二改造的i型crispr-cas效应子复合体结合dsdna中的第二核酸靶序列,cas3切口酶蛋白与第二复合体相关,并切割dsdna的第二链。改造的i型crispr-cas效应子复合体与相关的cas3切口酶蛋白的结合导致多核苷酸(例如,dsdna)中的核酸靶序列被改造的i型crispr-cas效应子复合体切割。实施例25a、实施例25b和实施例25c示出的数据证明了包含cas3 atp酶缺陷型突变体蛋白的cascade rnp复合体能够通过成对的切口诱导靶向的基因组缺失。该成对的切口可以促进宿主细胞(例如,人类细胞)基因组中的靶向缺失。
[0379]
在另一实施方案中,将包含与多核苷酸中的核酸靶序列互补的向导和第一核酸酶域(例如,foki)的改造的i型crispr-cas效应子复合体(图16a,cascade;虚线轮廓的框,经连接子多肽连接,黑色曲线,至第一核酸酶域,表示为圆扇形),以及能够与复合体结合的dcas3*-第二核酸酶域(例如,foki)融合蛋白(图16a,dcas3;实线轮廓的框,经连接子多肽连接,黑色曲线,至第二核酸酶域,表示为圆扇形)引入细胞或生化反应中。改造的i型crispr-cas效应子复合体(图16b,cascade)结合dsdna中的核酸靶序列(图16b,成对的水平黑线)并切割dsdna的第一链(图16c,cascade),并且dcas3*融合蛋白与cascade rnp复合体相关(图16b,dcas3*),并切割dsdna第二链(图16c,dcas3*)。
[0380]
在其他实施方案中,将包含与包含多核苷酸中的核酸靶序列的靶区互补的向导和能够与复合体结合的cas3蛋白(例如,cas3蛋白或mcas3蛋白)的改造的i型crispr-cas效应子复合体引入细胞或生化反应中。改造的i型crispr-cas效应子复合体结合dsdna中的核酸
靶序列,cas3蛋白(例如,cas3蛋白或mcas3蛋白)与复合体相关,并切割靶区中dsdna的至少一条链。在一些实施方案中,dsdna被mcas3蛋白切割导致dsdna靶区中的缺失。该方法可用于制作特定长度的长范围缺失,并且可以用于产生基因敲出或敲入。在一些实施方案中,cas3蛋白(例如,cas3蛋白或mcas3蛋白)可以融合至cascade复合体亚基蛋白(例如,cas7蛋白、cas8蛋白、cas5蛋白、cse2蛋白)。实施例23a至实施例23c描述了mcas3蛋白的实施方案。
[0381]
在另一实施方案中,本发明涉及使用i型crispr-cas效应子复合体,其中核酸酶域被融合至cascade复合体蛋白(参见,例如实施例11a,表38)或dcas3*蛋白(例如,融合至dna酶的dcas3*蛋白),以缺失核酸靶序列。该方法可用于在dsdna靶区中制作切割以及缺失,并且可以用于产生基因敲出。在一些实施方案中,可以将核酸酶域融合至cascade复合体亚基蛋白如cas7蛋白、cas8蛋白、cas5蛋白、cse2蛋白。
[0382]
切割多核苷酸中的核酸靶序列的方法还可以包括将供体多核苷酸引入细胞中,以促进将至少一部分的供体多核苷酸掺入细胞的gdna中。
[0383]
图17a示出了可被包含与多核苷酸中的第一核酸靶序列互补的向导(图17a,cascade1)和第一核酸酶域(例如,foki)的第一改造的i型crispr-cas效应子复合体(图17a,连接子多肽,示出为连接cascade1的弯曲线,和灰色的圆扇形),以及包含与多核苷酸中的第二核酸靶序列互补的向导(图17a,cascade 2)和第二核酸酶域(例如,foki)的第二改造的i型crispr-cas效应子复合体(图17a,连接子多肽,示出为连接cascade2的弯曲线,和灰色的圆扇形)切割的dsdna的两条链(图17a,成对的黑色水平线)的实例。图17b示出了包含与临近双链切割位点的dna序列互补的同源臂(图18b,供体,虚线)的供体多核苷酸(图17b,成对的虚线,显示在cascade2上方)。图17c示出了在双链切割位点区域中掺入的一部分的供体多核苷酸(图17c,连接代表dsdna的成对的黑色水平线的成对的虚线)。通过细胞dna修复机制(例如,hdr)介导供体多核苷酸的掺入(图17b至图17c,指向下,垂直箭头代表细胞dna修复机制)。
[0384]
在其他实施方案中,包含与多核苷酸中的第一核酸靶序列互补的向导和第一核酸酶域的改造的i型crispr-cas效应子复合体可以与包含第二核酸酶域的第二组分配对,其中第二组分能够结合多核苷酸的第二核酸靶序列。此类第二组分的实例包括包含第二核酸酶域的转录活化剂样效应子核酸酶(talen)、包含第二核酸酶域的锌指核酸酶(zfn),或者包含第二核酸酶域的dcas9/natna复合体。
[0385]
在一个实施方案中,可以使用包含与靶标多核苷酸中的第一核酸靶序列互补的向导的cascade复合体和dcas9/natna复合体的组合来缺失靶标多核苷酸(例如,gdna)的区域,其中natna包含与靶标多核苷酸中的第二核酸靶序列互补的间隔区序列。选择了第一和第二核酸靶序列以位于靶向用于缺失的核酸靶序列两侧。包含活性核酸内切酶活性的cas3蛋白与cascade复合体结合,然后渐进性地缺失包含靶向用于缺失的核酸靶序列的dsdna的单链。当cas3蛋白与dcas9/natna复合体(即“路障”)碰撞时,可以通过dcas9/natna复合体在第二核酸靶序列处终止cas3核酸酶活性。图21a-图21d示出了核酸靶序列的cas3缺失的实例。图21a显示了包含位于靶向用于缺失的核酸靶序列两侧的核酸靶序列1(图21a,nats1)和核酸靶序列2(图21a,nats2)的dsdna(图21a,成对的水平黑线)。图21a显示了cascade复合体,其包含与nats1互补的向导(图21a,cascade;黑线框架的矩形)、cas3蛋白(图21a,cas3;灰色圆扇形)和包含与nats2互补的间隔区的dcas9/natna复合体(图21a,
dcas9;虚线框架的矩形)。图21b显示了cascade复合体与nats1的结合、cas3蛋白与cascade复合体的关联,以及dcas9/natna复合体与nats2的结合。图21c示出了通过靶向用于缺失的单链核酸靶序列的cas3的渐进性缺失。图21d显示了在结合至nats2的dcas9/natna复合体的位置处cas3蛋白与dsdna的解离。实施例24a-实施例24d示出的数据支持了使用蛋白路障控制通过与cascade核蛋白复合体相关的cas3蛋白介导的缺失的长度;从而提供了使用与cascade核蛋白复合体相关的cas3蛋白来促进在细胞(例如,人类细胞)的gdna中形成具有限定长度的缺失的方法。
[0386]
在另一实施方案中,可以使用包含与靶标多核苷酸中的第一核酸靶序列互补的向导的第一cascade复合体和包含与靶标多核苷酸中的第二核酸靶序列互补的向导的第二cascade复合体的组合来缺失靶标多核苷酸(例如,gdna)的区域。选择了第一和第二核酸靶序列以位于靶向用于缺失的核酸靶序列的两侧。包含活性核酸内切酶活性的cas3蛋白具每种cascade复合体结合,然后渐进性地缺失靶向用于缺失的核酸靶序列的两条链。当每种cas3蛋白与一种cascade复合体碰撞时,可以在通过cascade复合体第一和第二核酸靶序列处终止cas3核酸酶活性。图22a-图22d示出了核酸靶序列的两条链的cas3缺失的实例。图22a显示了包含位于靶向用于缺失的核酸靶序列两侧的核酸靶序列1(图22a,nats1)和核酸靶序列2(图22a,nats2)的dsdna(图22a;成对的水平黑线)。图22a显示了第一cascade复合体,其包含与nats1互补的向导(图22a,cascade1;黑线框架的矩形)、cas3蛋白(图22a,cas3;灰色圆扇形),以及包含与nats2互补的向导的第二cascade复合体(图22a,cascade2;虚线框架的矩形)。图22b显示了cascade复合体与nats1和nats2的结合,以及cas3蛋白与cascade复合体的关联。图22c示出了由沿dna的移动和通过靶向用于缺失的核酸靶序列的两条链的cas3的核酸酶降解引起的渐进性的缺失。图22d显示了在与nats1和nats2结合的cascade复合体的位置处cas3蛋白与dsdna的解离。
[0387]
在其他实施方案中,可以修饰cascade复合体,使其不能与cas3蛋白结合,并且这种修饰的cascade复合体可以基本上以与图21a-图21d所示相同的方式用作路障,以通过催化活化与cascade rnp复合体相关的cas3来阻止dna的渐进性降解。另外的位点特异性结合蛋白(例如,转录活化剂样效应子(tal)或锌指(znf)dna结合蛋白)可以类似的方式用作路障。
[0388]
在一些实施方案中,核酸靶序列是dsdna(例如,基因组)dna。在一些实施方案中,核酸靶序列是双链的,并且一条或两条链被切割。此类切割核酸靶序列的方法可以在体外、体内或离体下进行。
[0389]
如上所述,在一些实施方案中,本发明涉及引入一种或多种改造的i型crispr-cas效应子复合体至宿主细胞中,以促进在供体多核苷酸存在下dsdna中核酸靶序列的切割,其中一种或多种改造的i型crispr-cas效应子复合体在包含宿主细胞dna的核酸靶序列的靶区中产生切割位点(或切割位点和相关的缺失),从而促进将至少一部分的供体多核苷酸插入靶区中。在一些实施方案中,切割位点是靶区中的双链断裂(例如,当使用每种包含间隔区的两种改造的i型crispr-cas效应子复合体以及包含cas蛋白和核酸内切酶(例如,foki)的融合蛋白或者每种包含与cas3蛋白或mcas3蛋白相关的间隔区的两种改造的i型crispr-cas效应子复合体时)。在一些实施方案中,切割位点是靶区中的单链断裂(例如,当使用与mcas3蛋白相关的i型crispr-cas效应子复合体时)。在其他实施方案中,切割位点是靶区中
的缺失(例如,当使用与cas3或mcas3蛋白相关的i型crispr-cas效应子复合体时)。
[0390]
为了证明同源性定向修复(hdr),设计了最小crispr阵列,以将foki-psecascade rnp复合体靶向人类基因组中的四个位点(wdr92、b2m、ccr5和trac)。使用三种寡核苷酸(seq id no:1513至seq id no:1515;实施例20a)和编码“重复区-间隔区-重复区-间隔区-重复区”序的独特引物,利用基于pcr的组装产生了最小crispr阵列,其中第一和第二间隔区将foki-psecascade rnp复合体引导至相邻的核酸靶序列,以使得能够进行foki二聚化和基因组切割(即产生切割位点)。
[0391]
对于包含切割位点的靶区中的每个hdr插入位点—在这种情况下与切割位点重叠,将细胞进行以下转染:编码包含其中nls与foki的n端连接的融合至cas8的n端的foki的foki-psecascade复合体蛋白组分的3μg的载体、150ng的最小crispr阵列,以及0-60pmol的hdr的单链寡脱氧核苷酸(ssodn)模板供体多核苷酸。ssodn包含同源臂,每个同源臂为75个核苷酸,并且两个臂对称地位于切割位点周围。供体多核苷酸在同源臂的3'端核苷酸处还包含硫代磷酸酯键,以减少或防止供体多核苷酸的细胞降解。在磷酸硫酯键的5'端,供体多核苷酸还包含“taataat”的插入序列,以插入两个终止密码子,并增加修复的染色体中的间隔区间距,从而阻碍foki-psecascade rnp复合体重新切割。
[0392]
基本上按实施例20b中所述在hek293细胞中进行转染,除了在混合物中包括ssodn以使得能够进行hdr。转染几天后,从细胞中纯化出gdna,用核酸外切酶进行处理以去除可能污染后续pcrs的任何残留ssodn,然后用作扩增的模板以测量供体插入。基本上按实施例20c中所述进行深度测序分析。表13中示出了来自该实验的总读长中突变体读长的百分比(第一栏是pmol的ssodn):
[0393][0394]
突变体读长的百分比指示了包含产生于非同源的末端连接以及“taataat”hdr序列的插入的插入缺失的突变体读长。
[0395]
在表14中示出了来自该实验的总突变体读长中仅含有“taataat”插入序列的hdr读长的百分比(第一栏是pmol的ssodn):
[0396]
[0397][0398]
如从数据可以看出的,通过cascade rnp复合体的dsdna切割使得能够在人类基因组中的多个基因座处进行hdr以及掺入供体多核苷酸。
[0399]
在又一个实施方案中,本发明包括在细胞或生化反应中修饰多核苷酸(例如,dna)中的一种或多种核酸靶序列的方法,包括提供一种或多种用于引入细胞或生化反应中的改造的i型crispr-cas效应子复合体(例如,包含cas亚基蛋白-胞苷脱氨酶融合蛋白),以及将改造的i型crispr-cas效应子复合体引入细胞或生化反应中,从而促进改造的i型crispr-cas效应子复合体与多核苷酸的接触,导致有利于核酸靶序列的突变(例如,c至t、g至a、a至g和t至c)的改造的i型crispr-cas效应子复合体与多核苷酸中的核酸靶序列的结合。图18a-图18d示出了使用包含cas亚基蛋白-连接子多肽-胞苷脱氨酶融合蛋白(cascade/cd复合体)的cascade复合体来突变细胞gdna中的靶标核苷酸(图18a,成对的黑色水平线,“c”为胞嘧啶且“g”为鸟嘌呤)的实例。将cascade/cd复合体(图18a;具有连接子多肽的“cascade”示出为连接cascade和胞苷脱氨酶的弯曲线,“cd”表示为灰色的圆扇形)引入细胞中。cascade/cd复合体包含与临近靶标胞嘧啶的dna靶序列互补的向导(图18b,“c”)。在图18b中,cascade/cd复合体结合dna靶序列,且胞苷脱氨酶将胞嘧啶(图18b,“c”)转化为胞嘧啶(图18c,“u”)。然后细胞修复机制可以将胞嘧啶修复成胸腺嘧啶,并将错配的胍改变为腺嘌呤(图18c-图18d,指向下,垂直箭头代表细胞dna修复机制)。
[0400]
在又一个实施方案中,本发明包括调节体外或体内转录,例如,包含调控元件序列的基因的转录的方法。这样的方法包括提供提供用于引入细胞或生化反应中的一种或多种改造的i型crispr-cas效应子复合体(例如,包含cas亚基蛋白-转录因子融合蛋白),以及将改造的i型crispr-cas效应子复合体引入细胞或生化反应中,从而促进改造的i型crispr-cas效应子复合体与调控元件序列的接触,导致改造的i型crispr-cas效应子复合体与调控元件序列结合,从而促进调节包含调控元件序列的基因的体外或体内转录。
[0401]
图19a和图19b示出了通用基因(“基因1”)的转录激活的实例的一般性图示。图19a提供了真核细胞中内源基因的转录调控的概述。在图19a中,两条黑色的平行线代表双链dna,指示了基因1(图19a,基因1)的位置,以及与基因1相关的转录起始位点(图19a,tss)。在图19a的第一幅图中,基因1的转录激活所需的转录因子(图19a,tf)和聚合酶ii(图19a,pol ii)被示出为尚未与基因1-tss关联。第二幅图示出了tf与其同源tss的关联。然后tf募集转录激活蛋白(图19a,tp),其随后募集rna聚合酶ii(图19a,pol ii)。通常,在真核生物中,tf因子和tp形成包含多种蛋白质和可能的其他分子的复合体。第三幅图示出了通过pol ii产生的对基因1的转录(图19a,基因1末端的弯曲箭头指示转录的方向)。这种类型的转录激活通常取决于特定于基因表达的tf。图19b示出了本发明的一个实施方案的图示,其中cascade复合体被改造以包括蛋白质或因子(图19b,cascadea),其吸引细胞中负责转录激
活的一种或多种组分(转录激活因子;图19b,ta)。这样的蛋白质或因子的一个实例是蛋白质vp64。cascadea包含能够在tss处或附近结合的向导(图19b,tss)。在图19b中,两条黑色平行线代表双链dna,指示了基因1(图19b,基因1)的位置,以及与基因1相关的转录起始位点(tss)。在图19b的第一幅图中,cascadea和聚合酶ii(图19b,pol ii)被示出为尚未与基因1-tss关联。在第二幅图中,示出了cascadea与其靶标tss的关联。然后cascadea募集转录激活蛋白(图19b,ta),其然后募集rna聚合酶ii(图19b,pol ii)。第三幅图示出了通过pol ii产生的对基因1的转录(图19b,基因1末端的弯曲箭头指示转录的方向)。本发明的该实施方案的一个优点是,基因的转录激活不依赖于与基因的tss结合的内源转录因子,而是可以通过选择适当的cascade向导来靶向基因的tss。
[0402]
图20a和图20b示出了使用包含cas亚基蛋白-krab域融合和与基因1相关的调控序列(图20a,启动子)互补的向导(图20a,具有示出为连接cascade和代表krab域的圆形元件的弯曲线的连接子多肽的cascadei)的cascade复合体,对通用基因(图20a,基因1)的转录抑制的实例的一般性图示。cascadei与调控序列的结合(图20b)导致基因1的转录抑制(图20b,以x结尾的黑线代表转录抑制)。
[0403]
可将如本文所述的改造的i型crispr-cas效应子复合体整合到试剂盒中。在一些实施方案中,试剂盒包括具有一个或多个容纳试剂盒元件的容器的包装,所述试剂盒元件作为一种或多种单独的组合物,或者,任选地,如果组分的相容性允许,则作为混合物。在一些实施方案中,试剂盒还包含一种或多种以下赋形剂:缓冲液、缓冲试剂、盐、无菌水溶液、防腐剂及以上的组合。示例性的试剂盒可包含一种或多种改造的i型crispr-cas效应子复合体和一种或多种赋形剂,或编码改造的i型crispr-cas效应子复合体的一种或多种组分的一种或多种核酸序列。
[0404]
此外,试剂盒还可以包括使用改造的i型crispr-cas效应子复合体组合物的说明书。
[0405]
本发明的另一个方面涉及制备或生产一种或多种改造的i型crispr-cas效应子复合体或其组分的方法。在一个实施方案中,制备或生产方法包括在细胞中产生改造的i型crispr-cas效应子复合体,以及从细胞溶解产物中纯化改造的i型crispr-cas效应子复合体。
[0406]
改造的i型crispr-cas效应子复合体组合物还可以包含可检测的标记,如可以提供可检测的信号的部分。可检测的标记的实例包括但不限于酶、放射性同位素、多种特异性结合对、荧光团(fam)、荧光蛋白(绿色荧光蛋白(gfp)、红色荧光蛋白、mcherry、tdtomato)、连同合适的荧光团(增强的gfp(egfp),“spinach”)一起的dna或rna适配体、量子点、抗体等。大量且各种合适的可检测的标记是本领域普通技术人员熟知的。
[0407]
在一些实施方案中,可以通过以下方法将改造的i型crispr-cas效应子复合体(即核蛋白颗粒)引入细胞中,这些方法包括但不限于:核转染、基因枪递送、声纳穿孔、细胞挤压、脂质转染,或使用其他化学物质、穿透细胞的肽等。在其他实施方案中,可使用载体系统、包含编码一种或多种组分的dna序列的表达盒,以及包含含有编码一种或多种组分的rna序列的表达盒的一种或多种rna分子(例如,mrna),将改造的i型crispr-cas效应子复合体和相关的蛋白质的一种或多种组分的编码序列引入细胞中。
[0408]
本发明的一个实施方案涉及使用改造的i型crispr-cas效应子复合体产生重组细
胞(例如,修饰的淋巴细胞)。方法通常包括促进使包含宿主细胞中的含有核酸靶序列的靶区的dsdna与本发明的一种或多种改造的第一类i型crispr-cas效应子复合体接触。改造的第一类i型crispr-cas效应子复合体与核酸靶序列的接触导致改造的第一类i型crispr-cas效应子复合体与包含核酸靶序列的靶区结合,切割包含核酸靶序列的靶区,以及修饰靶区中的dsdna,从而产生重组细胞。在一些实施方案中,dsdna包含一种以上的核酸靶序列,并且包含与每种核酸靶序列互补的间隔区序列的改造的第一类i型crispr-cas效应子复合体被用于结合、切割和修饰每种核酸靶序列。在一些实施方案中,靶区的修饰是插入、缺失或者插入和缺失。以上描述了切割多核苷酸中的核酸靶序列(例如dsdna中的单链切口或dsdna中的双链切口)的方法,包括提供用于引入细胞中的一种或多种改造的i型crispr-cas效应子复合体。
[0409]
本发明的实施方案包括使用一种或多种改造的第一类i型crispr-cas效应子复合体产生重组细胞,其中重组细胞的gdna包含敲出突变(例如,b2m基因和/或pdcd1基因的)、敲入(例如,trac位点处的编辑和来自供体多核苷酸的car的整合),或以上的组合。在一些实施方案中,在gdna的trac基因中核酸靶序列处的切割之后在核酸靶序列处掺入至少一部分的供体多核苷酸。供体多核苷酸可以包含car构建体,其中car被插入核酸靶序列中。
[0410]
通过本发明的方法制备的重组细胞可用于过继细胞转移(act)。act是一种迅速兴起的免疫疗法,其使用移植的免疫细胞来治疗癌症。act是将细胞转移到患者内。最常见的是,免疫细胞来源于免疫系统,旨在提高免疫功能。在自体癌症免疫疗法中,从患者内收获免疫细胞或干细胞,并通过离体培养至大数量进行扩增,然后返回患者内。免疫细胞或干细胞可以通过各种方式在培养中进行修饰(例如,使用基因组编辑将car整合到t细胞的基因组中)。在一些实施方案中,将用于修饰的淋巴细胞从对象中分离出来,进行修饰,然后重新引入同一对象中。这种技术被称为自体淋巴细胞疗法。在同种异体癌症免疫治疗中,源自单个供体的培养扩增的免疫细胞或干细胞可为大量患者提供治疗。这样的免疫细胞或干细胞也可以以各种方式在培养中进行修饰。在一些实施方案中,可以将淋巴细胞分离、修饰并引入不同的对象中。该技术被称为同种异体淋巴细胞疗法。
[0411]
在某些实施方案中,这种免疫治疗方法可以利用淋巴细胞,包括但不限于t细胞、自然杀伤细胞(nk细胞)、b细胞、肿瘤浸润淋巴细胞(til)、嵌合抗原受体t细胞(car-t细胞)、t细胞受体改造的t细胞(tcr)、tcr car-t细胞、car til细胞、car-nk细胞、改造的nk细胞或产生淋巴细胞的造血干细胞。在其他实施方案中,细胞是干细胞、树突状细胞等。此类细胞的基因组可以通过使用一种或多种本发明的改造的i类i型cascade效应子复合体来进行修饰(例如,淋巴细胞基因组中插入和/或缺失的产生)。
[0412]
可以从对象如人类对象,例如从血液或从实体瘤,如在tils的情况下,或从淋巴器官如胸腺、骨髓、淋巴结和粘膜相关的淋巴组织,分离淋巴细胞用于修饰。分离淋巴细胞的技术是本领域众所周知的。例如,可以从外周血单核细胞(pbmcs)中分离淋巴细胞,外周血单核细胞可以使用例如ficoll,一种分离血液层的亲水性多糖和密度梯度离心从全血中分离出来。通常,将抗凝或去纤维化的血液样本铺在ficoll溶液的顶部,并离心以形成不同的细胞层。底层包括红血球(红细胞),它们通过ficoll介质进行收集或聚集,并完全沉入底层。下一层主要包含粒细胞,它们也向下迁移通过ficoll-paque溶液。下一层包括淋巴细胞以及单核细胞和血小板,淋巴细胞通常位于血浆和ficoll溶液之间的界面处。为了分离淋
巴细胞,回收该层,用盐溶液洗涤以除去血小板、ficoll和血浆,然后再次离心。可选地,可以通过离心技术(例如,使用(haemonetrics,braintree,ma)机器或lovo自动化细胞处理系统(fresenius kabi usa,llc,lake zurich,il))从供体血液中分离细胞。
[0413]
用于分离淋巴细胞的其他技术包括生物淘选,其通过将目标细胞与抗体包被的塑料表面结合而从溶液中分离细胞群。然后通过用特异性抗体和补体处理去除不需要的细胞。另外,荧光活化细胞分选(facs)分析可用于检测和计数淋巴细胞。facs分析使用流式细胞仪,其可基于光散射和荧光的差异分离标记的细胞。
[0414]
对于tils,从肿瘤中分离淋巴细胞,并例如使其在大剂量il-2中生长,并使用针对自体肿瘤或hla匹配的肿瘤细胞系的细胞因子释放共培养测定进行选择。选择与同种异体的非mhc匹配的对照相比具有增加的特异性反应性的证据的培养物进行快速扩增,然后将其引入对象中以治疗癌症(参见,例如rosenberg,s.,et al.,clin.cancer res.17:4550-4557(2011);dudly,m.,et al.,science 298:850-854(2002);dudly,m.,et al.,j.clin.oncol.26:5233-5239(2008);dudley,m.,et al.,j.immnother.26:332-342(2003))。
[0415]
分离后,淋巴细胞可以根据特异性、频率和功能来进行定征。经常使用的测定包括elispot测定,其可测量t细胞反应的频率。
[0416]
在一些实施方案中,cd4+和cd8+t细胞是从供体外周血单核细胞(pbmcs)中分离出来的。本领域普通技术人员可以通过如上所述的各种方法分离t细胞或其他淋巴样细胞。还可通过自ipsc细胞的分化来分离此类细胞。
[0417]
分离后,可以使用本领域已知的技术活化淋巴细胞,以促进增殖和分化为特化的效应子淋巴细胞。活化的t细胞的表面标记包括例如cd3、cd4、cd8、pd1、il2r和其他。活化的细胞毒性淋巴细胞可在结合靶标细胞表面上的同源受体后杀伤靶标细胞。nk细胞的表面标记包括例如cd16、cd56和其他。
[0418]
在分离和任选地活化后,可以修饰淋巴细胞以提供所需的特性。本发明的一种或多种改造的i型cascade效应子复合体可以用于引入基因组修饰,包括但不限于引入待表达的编码序列和/或使内源基因表达失活。在一些实施方案中,本发明的一种或多种改造的i型cascade效应子复合体可用于编辑trac基因(编码t细胞受体α常数)、b2m基因(编码β2微球蛋白),和/或pdcd1基因(编码程序化细胞死亡蛋白1;也称为pd-1)。
[0419]
t细胞和nk细胞是可以通过本发明的方法修饰的淋巴细胞的实例。在一些实施方案中,本发明的一种或多种改造的i型cascade效应子复合体可以在存在包含car的供体多核苷酸的情况下,在基因的靶区中引入切割位点,其中car被整合到淋巴细胞基因组的靶区中。在另外的实施方案中,本发明的一种或多种改造的i型cascade效应子复合体可以用于在基因的靶区中引入切割位点,以促进敲出突变的产生,以防止基因的表达。
[0420]
在另一个实施方案中,可以使用本发明的改造的i型cascade效应子复合体将基因组修饰引入人ipscs中。在一些实施方案中,本发明的一种或多种改造的i型cascade效应子复合体可用于编辑trac基因、b2m基因和/或pdcd1基因。在其他实施方案中,改造的i型cascade效应子复合体与供体多核苷酸一起可用于引入基因组修饰和编码序列,如car或细胞因子(例如,il2,il15等)。然后可以将修饰的ipsc细胞进一步分化为包含t细胞和nk细胞或树突状细胞的成熟细胞类型。在一些实施方案中,修饰的ipscs可以分化为car-t细胞和
car-nk细胞。
[0421]
在本发明的方法的一些实施方案中,供体多核苷酸包含编码car的多核苷酸。可以对car进行靶向,以经由同源重组(“敲入”)插入包含切割位点的基因(例如,trac基因)的靶区中。这种方法的一个优点是它还可以提供靶向的trac基因的敲出;也就是说,使得trac基因失效。上文描述了可以掺入car构建体中的细胞外抗原识别域的实例(参见表2)。在一个实施方案中,细胞外抗原识别域包含cd19结合部分(例如,抗cd19 scfv)。在另一个实施方案中,细胞外抗原识别域包含b细胞成熟抗原(bcma)结合部分(例如,抗bcma scfv)。
[0422]
在本发明的包括在dna的靶区中产生切割位点的方法的实施方案中,方法还可以包括将供体多核苷酸引入修饰的细胞中,从而促进至少一部分供体多核苷酸的插入包含修饰细胞的切割位点的靶区中。可以将供体多核苷酸直接引入修饰的细胞中。在一些实施方案中,使用载体引入供体多核苷酸。用于构建载体的一般方法是本领域已知的。病毒载体的实例包括但不限于慢病毒、逆转录病毒、腺病毒、单纯疱疹病毒i或ii、细小病毒、网状内皮增生病毒和aav载体。
[0423]
本发明方法的其他实施方案包括在b2m基因中引入突变。在优选的实施方案中,突变是b2m基因中的敲出突变。
[0424]
本发明方法的其他实施方案包括在pdcd1基因中引入突变。在优选的实施方案中,突变是pdcd1基因中的敲出突变。
[0425]
可以通过同时或连续引入改造的cascade复合体、多核苷酸(例如,质粒或表达盒)或以上的混合物至宿主细胞(例如,淋巴细胞)中来进行被本发明的一种或多种改造的i型cascade效应子复合体促进的基因组修饰。
[0426]
在产生修饰的淋巴细胞后,可以筛选淋巴细胞,以使用诸如高通量筛选技术的方法,包括但不限于facs、基于微流体的筛选平台等选择表达(例如,表达所需的细胞表面受体)或不表达(例如,细胞表面蛋白,其表达已经通过使用一种或多种改造的i型cascade效应子复合体的基因组编辑进行了失活)的细胞。这些技术在本领域中是已知的(参见,例如wojcik,m.,et al.,int.j.mol..sci.16:24918-24945(2015))。
[0427]
修饰的淋巴细胞一旦产生,就可以配制成药物组合物,以递送至待治疗的对象。本发明的组合物包括修饰的淋巴细胞和一种或多种药学上可接受的赋形剂。示例性的赋形剂包括但不限于碳水化合物、无机盐、抗微生物剂、抗氧化剂、表面活性剂、缓冲剂、酸、碱及以上的组合。适合用于可注射组合物的赋形剂包括水、醇、多元醇、甘油、植物油、磷脂和表面活性剂。碳水化合物如糖、衍生化的糖如糖醇、醛糖酸、酯化的糖和/或糖聚合物可以作为赋形剂提供。特定的碳水化合物赋形剂包括,例如:单糖,如果糖、麦芽糖、半乳糖、葡萄糖、d-甘露糖、山梨糖等;二糖,如乳糖、蔗糖、海藻糖、纤维二糖等;多糖,如棉子糖、松三糖、麦芽糖糊精、右旋糖酐、淀粉等;以醛糖醇,如甘露醇、木糖醇、麦芽糖醇、乳糖醇、木糖醇、山梨糖醇(葡萄糖醇)、吡喃糖基山梨糖醇、肌醇等。赋形剂还可包括无机盐或缓冲剂,如柠檬酸、氯化钠、氯化钾、硫酸钠、硝酸钾、磷酸二氢钠、磷酸氢二钠及以上的组合。冷冻剂(例如,(biolife solutions inc,bothell,wa)cs2、cs5或cs10冷冻培养基)可用于冷冻细胞以进行存储和运输。
[0428]
本发明的药物组合物还可以包括用于防止或阻止微生物生长的抗微生物剂。适用于本发明的抗微生物剂的非限制性实例包括苯扎氯铵、苄索氯铵、苄醇、十六烷基氯化吡啶
鎓、氯丁醇、苯酚、苯乙醇、硝酸苯汞、硫柳汞及以上的组合。
[0429]
药物组合物中也可以存在抗氧化剂。抗氧化剂用于防止氧化,从而防止制剂中淋巴细胞或其他组分的变质。适合用于本发明的抗氧化剂包括例如抗坏血酸棕榈酸酯、丁基化羟基茴香醚、丁基化羟基甲苯、次磷酸、单硫代甘油、没食子酸丙酯、亚硫酸氢钠、甲醛合次硫酸氢钠、偏亚硫酸氢钠及以上的组合。
[0430]
表面活性剂可以作为赋形剂存在。示例性的表面活性剂包括:聚山梨酸酯,如tween 20和tween 80,以及普朗尼克类化合物,如f68和f88(basf,mount olive,new jersey);山梨糖醇酯;脂质,如磷脂,如卵磷脂和其他磷脂酰胆碱、磷脂酰乙醇胺(尽管优选不为脂质体形式)、脂肪酸和脂肪酯;类固醇,如胆固醇;螯合剂,如edta;以及锌和其他此类合适的阳离子。
[0431]
酸或碱可以作为药物组合物中的赋形剂存在。可以使用的酸的非限制性实例包括选自以下的那些酸:盐酸、乙酸、磷酸、柠檬酸、苹果酸、乳酸、甲酸、三氯乙酸、硝酸、高氯酸、磷酸、硫酸、富马酸及以上的组合。合适的碱的实例包括但不限于选自以下的碱:氢氧化钠、乙酸钠、氢氧化铵、氢氧化钾、乙酸铵、乙酸钾、磷酸钠、磷酸钾、柠檬酸钠、甲酸钠、硫酸钠、硫酸钾、富马酸钾及以上的组合。
[0432]
组合物中淋巴细胞(或其他重组细胞)的数量将根据多种因子而变化,但是当组合物处于单位剂量形式或容器(例如,袋)中时,其将最佳地为治疗有效剂量。治疗有效剂量可以通过重复给予增加量的组合物来实验确定,以确定哪种量产生临床所需的终点。
[0433]
组合物中任何单一赋形剂的量将根据赋形剂的性质和功能以及组合物的特定需要而变化。通常,任何单一赋形剂的最佳量是通过常规实验确定的,即通过制备包含不同量的赋形剂(范围从低到高)的组合物,检查稳定性和其他参数,然后确定取得最佳性能且无明显副作用的范围。然而,通常,赋形剂在组合物中存在的量为按重量计约1%至约99%,优选按重量计约5%至约98%,更优选按赋形剂重量计约15%至约95%,按重量计浓度小于30%是最优选的。这些上述的药物赋形剂连同其他的赋形剂描述于“remington:the science&practice of pharmacy,”现行版,williams&williams;the“physician
’
s desk reference,”现行版,medical economics,montvale,nj;和kibbe,a.h.,handbook of pharmaceutical excipients,现行版,american pharmaceutical association,washington,d.c.中。
[0434]
可以根据递送和使用的预期方式将药物组合物容纳在注射器、植入装置等中。优选地,存在的组合物的量适合于单一剂量,采用预先测量或预先包装的形式。
[0435]
本文中的药物组合物可以任选地包括一种或多种另外的试剂,如用于治疗对象的目标癌症或治疗该治疗的已知副作用的其他药物。例如,t细胞将细胞因子释放到血液中,这可能导致危险的高烧和血压急剧下降。这种病况称为细胞因子释放综合征(crs)。在许多患者中,crs可以通过标准的支持疗法进行管理,包括类固醇和免疫疗法,如能阻断il-6活性的托珠单抗(actemratm,genentech,south san francisco,ca)。
[0436]
至少一个治疗有效周期的修饰的淋巴细胞组合物治疗将被给予对象。通过“治疗有效周期的治疗”意指这样一种治疗周期,当给予该治疗周期时,其引起关于个体目标疾病治疗的积极的治疗反应。通过“积极的治疗反应”意指根据本发明进行治疗的个体表现出疾病的一种或多种症状的改善,包括诸如肿瘤减少和/或对淋巴细胞治疗的需求减少的改善。
[0437]
在某些实施方案中,将给予多种治疗有效剂量的包含淋巴细胞或其他药物的组合物。本发明的组合物,尽管不一定,但通常是经注射,如皮下、皮内、静脉内、动脉内、肌内、腹膜内、髓内、肿瘤内、结节内),通过输注或局部给药的。在即将给药前,药物制剂可以为液体溶液或悬浮液的形式。上述内容旨在是示例性的,因为还考虑了另外的给药方式。可以根据本领域已知的任何医学上可接受的方法,使用相同或不同的给药途径来给予药物组合物。
[0438]
实际给予的剂量将根据对象的年龄、体重和一般状况以及所治疗病况的严重性、医疗保健专业人员的判断以及所给予的特定淋巴细胞而变化。治疗有效量可以由本领域技术人员确定,并将根据每种特定情况的特定要求调整。
[0439]
通常,每位患者淋巴细胞的治疗有效量范围将为总计约1x105个至约1x10
10
个淋巴细胞或更多,如1x106个至约1x10
10
个,例如,1x107个至1x109个,如5x107个到5x108个,或这些范围内的任何数量。其他剂量范围可以是每kg/体重1x104个至1x10
10
个细胞。淋巴细胞的总数可以单次大剂量给药,也可以分两次或更多次剂量给药,如相隔一天或多天。淋巴细胞的总数可以单次大剂量给药,或者可以分两次或更多次剂量给药,如间隔一天或更多天。给予的化合物的量将取决于特定淋巴细胞组合物的效力、所治疗的疾病和给药途径。
[0440]
另外,剂量可以包括淋巴细胞的混合物,如cd8+和cd4+细胞的混合物。如果提供了cd8+和cd4+细胞的混合物,则cd8+与cd4+细胞的比例可以为例如1:1、1:2或2:1、1:3或3:1、1:4或4:1、1:5或5:1等。
[0441]
修饰的淋巴细胞可以在其他试剂之前、同时或之后给药。如果与其他试剂同时提供,则修饰的淋巴细胞可以在相同或不同的组合物中提供。因此,可以通过同时治疗的方式将淋巴细胞和其他试剂提供给个体。通过“同时治疗”,意指给予对象,使得在接受治疗的对象中引起物质组合的治疗效果。例如,根据特定的给药方案,可以通过给予组合包含治疗有效剂量的一剂包含修饰的淋巴细胞的药物组合物和一剂包含至少一种其他的试剂,如另一化疗剂的药物组合物来实现同时治疗。类似地,可以在至少一次治疗剂量中给予修饰的淋巴细胞和治疗剂。可以同时或在不同时间(例如,连续地、以任一顺序,在同一天或在不同天)进行单独药物组合物的给药,只要在接受治疗的对象中引起了这些物质组合的治疗效果。
[0442]
如本文所述,本发明的改造的i型cascade效应子复合体提供了基因组编辑工具。证明第一类crispr-cas系统在用于基因组编辑的哺乳动物细胞中进行了功能重构的实验表明,此类改进的质粒设计能够允许使用其他的第一类crispr-cas系统,包括显示更少的蛋白组分和独特的pam要求的那些,以及可能地甚至来自iii型crispr
–
cas系统的rna和dna靶向性效应子复合体(参见,例如hille,f.,et al.,cell 172:1239-1259(2018);tamulaitis,g.,et al.,trends microbiol.25:49-61(2017))。cascade复合体的多亚基性质提供了多价和/或空间上精确募集效应子融合物的可能性,如合成的转录因子、表观基因组修饰剂和碱基编辑器。另外,可以利用异源表达来自i型系统的完整dna干扰途径
–
即cascade介导的募集cas3解旋酶-核酸酶至基因组靶标位点
–
以产生较大的dna缺失,来暴露较长的ssdna束用于同源指导的修复,和/或机械破坏确定的基因组位点处的蛋白-dna路障。因此,在本发明的一个实施方案中,改造的第一类crispr-cas系统可用于产生较大的缺失区域,并且可将供体多核苷酸(例如,比较合适的同源臂)引入细胞中,从而促进将至少一部分的供体多核苷酸插入该区域中。
[0443]
本发明的实施方案包括但不限于以下各项。
[0444]
实施方案1.组合物,其包含:
[0445]
第一改造的第一类i型crispr-cas效应子复合体,其包含:
[0446]
第一cse2亚基蛋白、第一cas5亚基蛋白、第一cas6亚基蛋白和第一cas7亚基蛋白,
[0447]
包含第一cas8亚基蛋白和第一foki的第一融合蛋白,其中第一cas8亚基蛋白的n端或第一cas8亚基蛋白的c端通过第一连接子多肽分别与第一foki的c端或n端共价连接,并且其中第一连接子多肽具有约10个氨基酸至约40个氨基酸的长度,和
[0448]
包含能够结合第一核酸靶序列的第一间隔区的第一向导多核苷酸;以及
[0449]
第二改造的第一类i型crispr-cas效应子复合体,其包含:
[0450]
第二cse2亚基蛋白、第二cas5亚基蛋白、第二cas6亚基蛋白和第二cas7亚基蛋白,
[0451]
包含第二cas8亚基蛋白和第二foki的第二融合蛋白,其中第二cas8亚基蛋白的n端或第二cas8亚基蛋白的c端通过第二连接子多肽分别与第二foki的c端或n端共价连接,并且其中第二连接子多肽具有约10个氨基酸至约40个氨基酸的长度,和
[0452]
包含能够结合第二核酸靶序列的第二间隔区的第二向导多核苷酸,其中第二核酸靶序列的前间区序列邻近基序(pam)和第一核酸靶序列的pam具有约20bp至约42bp的间隔区间距。
[0453]
实施方案2.如实施方案1所述的组合物,其中第一连接子多肽具有约15个氨基酸至约30个氨基酸的长度。
[0454]
实施方案3.如实施方案2所述的组合物,其中所述第一连接子多肽具有约17个氨基酸至约20个氨基酸的长度。
[0455]
实施方案4.如实施方案1-3中任一项所述的组合物,其中第二连接子多肽具有约15个氨基酸至约30个氨基酸的长度。
[0456]
实施方案5.如实施方案4所述的组合物,其中第二连接子多肽具有约17个氨基酸至约20个氨基酸的长度。
[0457]
实施方案6.如任何前述实施方案所述的组合物,其中第一连接子多肽和第二连接子多肽的长度是相同的。
[0458]
实施方案7.如任何前述实施方案所述的组合物,其中第二核酸靶序列和第一核酸靶序列中的每个具有约22bp至约40bp的间隔区间距。
[0459]
实施方案8.如实施方案7所述的组合物,其中第二核酸靶序列和第一核酸靶序列中的每个具有约26bp至约36bp的间隔区间距。
[0460]
实施方案9.如实施方案8所述的组合物,其中第二核酸靶序列和第一核酸靶序列中的每个具有约29bp至约35bp的间隔区间距。
[0461]
实施方案10.如实施方案9所述的组合物,其中第二核酸靶序列和第一核酸靶序列中的每个具有约30bp至约34个碱基bp的间隔区间距。
[0462]
实施方案11.如任何前述实施方案所述的组合物,其中第一foki和第二foki是能够结合形成同型二聚体的单体亚基。
[0463]
实施方案12.如实施方案1-10中任一项所述的组合物,其中第一foki和第二foki是能够结合形成异型二聚体的不同的单体亚基。
[0464]
实施方案13.如任何前述实施方案所述的组合物,其中第一cas8亚基蛋白的n端通
过第一连接子多肽与第一foki的c端共价连接。
[0465]
实施方案14.如实施方案1-12中任一项所述的组合物,其中第一cas8亚基蛋白的c端通过第一连接子多肽与第一foki的n端共价连接。
[0466]
实施方案15.如任何前述实施方案所述的组合物,其中第二cas8亚基蛋白的n端通过第二连接子多肽与第二foki的c端共价连接。
[0467]
实施方案16.如实施方案1-14中任一项所述的组合物,其中第二cas8亚基蛋白的c端通过第二连接子多肽与第二foki的n端共价连接。
[0468]
实施方案17.如任何前述实施方案所述的组合物,其中第一cas8亚基蛋白和第二cas8亚基蛋白中的每个包含相同的氨基酸序列。
[0469]
实施方案18.如任何前述实施方案所述的组合物,其中第一cse2亚基蛋白和第二cse2亚基蛋白中的每个包含相同的氨基酸序列,第一cas5亚基蛋白和第二cas5亚基蛋白中的每个包含相同的氨基酸序列,第一cas6亚基蛋白和第二cas6亚基蛋白中的每个包含相同的氨基酸序列,并且第一cas7亚基蛋白和第二cas7亚基蛋白中的每个包含相同的氨基酸序列。
[0470]
实施方案19.如任何前述实施方案所述的组合物,其中第一向导多核苷酸包含rna。
[0471]
实施方案20.如任何前述实施方案所述的组合物,其中第二向导多核苷酸包含rna。
[0472]
实施方案21.如任何前述实施方案所述的组合物,其中基因组dna包含第二核酸靶序列的pam和第一核酸靶序列的pam。
[0473]
实施方案22.细胞,其包含:任何前述实施方案所述的组合物。
[0474]
实施方案23.如实施方案22所述的细胞,其中细胞的基因组dna包含第二核酸靶序列的pam和第一核酸靶序列的pam。
[0475]
实施方案24.如实施方案22或23所述的细胞,其中细胞是原核细胞。
[0476]
实施方案25.如实施方案22或23所述的细胞,其中细胞是真核细胞。
[0477]
实施方案26.一种或多种核酸序列,其编码实施方案1-21中任一项所述的第一cse2亚基蛋白、第一cas5亚基蛋白、第一cas6亚基蛋白、第一cas7亚基蛋白、第一融合蛋白和第一向导多核苷酸。
[0478]
实施方案27.一种或多种核酸序列,其编码实施方案1-21中任一项所述的第二cse2亚基蛋白、第二cas5亚基蛋白、第二cas6亚基蛋白、第二cas7亚基蛋白、第二融合蛋白和第二向导多核苷酸。
[0479]
实施方案28.一种或多种表达盒,其包含实施方案26、实施方案27或实施方案26和实施方案27所述的一种或多种核酸序列。
[0480]
实施方案29.一种或多种载体,其包含实施方案28所述的一种或多种表达盒。
[0481]
实施方案30.结合包含第一核酸靶序列和第二核酸靶序列的多核苷酸的方法,所述方法包括:
[0482]
提供实施方案1-21中任一项所述的组合物,以用于引入细胞或生化反应中;以及
[0483]
将组合物引入细胞或生化反应中,从而促进第一改造的第一类i型crispr-cas效应子复合体与第一核酸靶序列的接触,以及第二改造的第一类i型crispr-cas效应子复合
体与第二核酸靶序列的接触,导致第一改造的第一类i型crispr-cas效应子复合体与第一核酸靶序列结合,以及第二改造的第一类i型crispr-cas效应子复合体与多核苷酸中的第二核酸靶序列结合。
[0484]
实施方案31.如实施方案30所述的方法,其中基因组dna包含多核苷酸。
[0485]
实施方案32.切割包含第一核酸靶序列和第二核酸靶序列的多核苷酸的方法,所述方法包括:
[0486]
提供实施方案1-21中任一项所述的组合物,以用于引入细胞或生化反应中;以及
[0487]
将组合物引入细胞或生化反应中,从而促进第一改造的第一类i型crispr-cas效应子复合体与第一核酸靶序列接触,以及改造的第二第一类i型crispr-cas效应子复合体与第二核酸靶序列接触,导致第一核酸靶序列被第一改造的第一类i型crispr-cas效应子复合体切割,以及第二核酸靶序列被第二改造的第一类i型crispr-cas效应子复合体切割。
[0488]
实施方案33.如实施方案32所述的方法,其中基因组dna包含多核苷酸。
[0489]
实施方案34.试剂盒,其包含:实施方案1-21中任一项所述的组合物;和缓冲剂。
[0490]
实施方案35.试剂盒,其包含:实施方案26、实施方案27或实施方案26和实施方案27所述的一种或多种核酸序列;和缓冲剂。
[0491]
实施方案36.组合物,其包含:
[0492]
改造的第一类i型crispr-cas效应子复合体,其包含:
[0493]
cse2亚基蛋白、cas5亚基蛋白、cas6亚基蛋白和cas7亚基蛋白,
[0494]
包含cas8亚基蛋白和第一foki的第一融合蛋白,其中第一cas8亚基蛋白的n端或第一cas8亚基蛋白的c端通过第一连接子多肽分别与第一foki的c端或n端共价连接,和
[0495]
包含能够结合核酸靶序列的间隔区的向导多核苷酸;以及
[0496]
第二融合蛋白,其包含含有dcas3*蛋白和第二foki的改造的第一类i型crispr-cas3融合蛋白,其中dcas3*蛋白的n端或dcas3*蛋白的c端通过第二连接子多肽分别与第二foki的c端或n端共价连接,并且其中第一连接子多肽具有约10个氨基酸至约40个氨基酸的长度,效应子复合体包含,
[0497]
实施方案37.如实施方案36所述的组合物,其中第一连接子多肽具有约5个氨基酸至约40个氨基酸的长度。
[0498]
实施方案38.如实施方案36所述的组合物,其中第二连接子多肽具有约5个氨基酸至约40个氨基酸的长度。
[0499]
实施方案39.细胞,其包含:实施方案36-38中任一项所述的组合物。
[0500]
实施方案40.如实施方案39所述的细胞,其中细胞是原核细胞。
[0501]
实施方案41.如实施方案39所述的细胞,其中细胞是真核细胞.
[0502]
实施方案42.一种或多种核酸序列,其编码实施方案36-38中任一项所述的cse2亚基蛋白、cas5亚基蛋白、cas6亚基蛋白、cas7亚基蛋白、第一融合蛋白和向导多核苷酸。
[0503]
实施方案43.一种或多种核酸序列,其编码实施方案36-38中任一项所述的第二融合蛋白。
[0504]
实施方案44.一种或多种表达盒,其包含实施方案42、实施方案43或实施方案42和实施方案43的一种或多种核酸序列。
[0505]
实施方案45.一种或多种载体,其包含实施方案44所述的一种或多种表达盒。
[0506]
实施方案46.结合包含核酸靶序列的多核苷酸的方法,所述方法包括:
[0507]
提供实施方案36-38中任一项所述的组合物,以用于引入细胞或生化反应中;以及
[0508]
将组合物引入细胞或生化反应中,从而促进改造的第一类i型crispr-cas效应子复合体与核酸靶序列的接触,以及第二融合蛋白与改造的第一类i型crispr-cas效应子复合体的接触,导致改造的第一类i型crispr-cas效应子复合体和第二融合蛋白与多核苷酸中的核酸靶序列结合。
[0509]
实施方案47.如实施方案46所述的方法,其中基因组dna包含多核苷酸。
[0510]
实施方案48.切割包含核酸靶序列的多核苷酸的方法,所述方法包括:
[0511]
提供实施方案36-38中任一项所述的组合物,以用于引入细胞或生化反应中;以及
[0512]
将组合物引入细胞或生化反应中,从而促进第一改造的第一类i型crispr-cas效应子复合体与第一核酸靶序列接触,以及改造的第二第一类i型crispr-cas效应子复合体与第二核酸靶序列接触,
[0513]
将组合物引入细胞或生化反应中,从而促进改造的第一类i型crispr-cas效应子复合体与核酸靶序列接触,以及第二融合蛋白与改造的第一类i型crispr-cas效应子复合体接触,导致核酸靶序列被改造的第一类i型crispr-cas效应子复合体和第二融合蛋白切割。
[0514]
实施方案49.如实施方案48所述的方法,其中基因组dna包含多核苷酸。
[0515]
实施方案50.试剂盒,其包含:实施方案36-38中任一项所述的组合物;和缓冲剂。
[0516]
实施方案51.试剂盒,其包含实施方案42、实施方案43或实施方案42和实施方案43所述的一种或多种核酸序列;和缓冲剂。
[0517]
实施方案52.相对于野生型i型crisprcas3蛋白(“wtcas3蛋白”),能够减少沿dna的移动的改造的i型crisprcas3突变体蛋白(“mcas3蛋白”),所述mcas3蛋白包含:
[0518]
与相应的wtcas3蛋白的约95%或更高的序列同一性,
[0519]
核定位信号在氨基端、羧基端或氨基端和羧基端处共价连接,以及
[0520]
能够下调解旋酶活性的一个或多个突变,其中改造的i型crisprcas3突变体蛋白保留了核酸酶活性;
[0521]
其中dna是包含含有核酸靶序列的靶区的双链dna(dsdna);
[0522]
其中当wtcas3蛋白与相应的cascade核蛋白复合体(“cascade np复合体/wtcas3蛋白”)关联,并且cascade np复合体包含含有与核酸靶序列互补的间隔区的向导时,cascade np复合体/wtcas3蛋白与核酸靶序列的结合有利于dna靶区中的切割,从而导致缺失(“wtcas3-缺失”);并且
[0523]
其中mcas3蛋白当其与cascade np复合体(“cascade np复合体/mcas3蛋白)关联并结合核酸靶序列时,有利于dna靶区中的切割,从而导致相对于wtcas3-缺失的更短的缺失。
[0524]
实施方案53.如实施方案53所述的mcas3蛋白,其中一个或多个突变是氨基酸的取代。
[0525]
实施方案54.如任何前述实施方案所述的mcas3蛋白,其中一个或多个突变在解旋酶域的reca1区域或reca2区域中。
[0526]
实施方案55.如任何前述实施方案所述的mcas3蛋白,其中一个或多个突变相对于
wtcas3蛋白下调mcas3蛋白与单链dna(ssdna)的结合。
[0527]
实施方案56.如任何前述实施方案所述的mcas3蛋白,其中,一个或多个突变下调三磷酸腺苷(atp)被mcas3蛋白的水解,或下调atp与mcas3的结合蛋白。
[0528]
实施方案57.如任何前述实施方案所述的mcas3蛋白,其中mcas3蛋白的编码序列共价连接至cascade np复合体的cas蛋白的编码序列的氨基端或羧基端。
[0529]
实施方案58.如任何前述实施方案所述的mcas3蛋白,其中一个或多个突变相对于wtcas3蛋白下调mcas3蛋白与单链dna(ssdna)的结合。
[0530]
实施方案59.如任何前述实施方案所述的mcas3蛋白,其中mcas3蛋白的编码序列共价连接至cascade rnp复合体的cas蛋白的编码序列的氨基端或羧基端。
[0531]
实施方案60.如任何前述实施方案所述的mcas3蛋白,其中cas蛋白选自:cse2、cas8蛋白、cas7蛋白、cas6蛋白和cas5蛋白。
[0532]
实施方案61.如任何前述实施方案所述的mcas3蛋白,其中wtcas3蛋白是大肠杆菌1类crisprcas3蛋白。
[0533]
实施方案62.如实施方案61方案所述的mcas3蛋白,其中一个或多个突变选自d452h、a602v以及d452h和a602v。
[0534]
实施方案63.如任何前述实施方案所述的mcas3蛋白,其中dna在细胞中。
[0535]
实施方案64.如实施方案63所述的mcas3蛋白,其中细胞是真核细胞。
[0536]
实施方案65.如实施方案64所述的mcas3蛋白,其中真核细胞是哺乳动物细胞(例如,人类细胞)。
[0537]
实施方案66.一种或多种多核苷酸,其编码实施方案52-65中任一项所述的mcas3蛋白。
[0538]
实施方案67.质粒,其包含编码实施方案52-65中任一项所述的mcas3蛋白的可操作地连接至调控序列以用于在哺乳动物细胞中表达的多核苷酸序列。
[0539]
实施方案68.一种或多种质粒,其包含编码实施方案52-65中任一项所述的mcas3蛋白的多核苷酸序列,以及编码相应的i型crisprcascade的蛋白组分的可操作地连接至调控序列以用于在哺乳动物细胞中表达的一种或多种多核苷酸。
[0540]
实施方案69.如实施方案68所述的一种或多种质粒,还包括编码可操作地连接至调控序列以用于在哺乳动物细胞中表达的一种或多种向导多核苷酸的质粒。
[0541]
实施方案70.i型crisprcascade核蛋白复合体,其包含实施方案52-65中任一项所述的mcas3蛋白。
[0542]
实施方案71.如实施方案70所述的i型crisprcascade核蛋白复合体,其中核蛋白复合体是rnp。
[0543]
尽管已经在本文中显示和描述了本发明的优选的实施方案,但是对于本领域技术人员而言显而易见的是,仅通过实例的方式来提供此类实施方案。从本说明书和实施例,本领域技术人员可以确定本发明的基本特征,并且在不脱离本发明的精神和范围的情况下,可以对本发明进行改变、替代、变更和修改,以使其适应各种用途和条件。这样的改变、替代、变更和修改也旨在落入本公开的范围内。
[0544]
实例
[0545]
在以下实施例中示出了本发明的方面。已经进行了努力以确保关于所使用的数字
(例如,数量、浓度、变化百分比等)的准确性,但应考虑一些实验误差和偏差。除非另有指示,否则温度为摄氏度,且压力为大气压或接近大气压。应当理解,这些实施例仅通过举例说明的方式给出,且并不旨在限制本发明的范围。
[0546]
实施例1
[0547]
编码cascade组分的多核苷酸的计算机设计
[0548]
本实施例提供了使用来源于i-e型crispr-cas系统的基因、蛋白和crispr序列设计编码cascade的多核苷酸组分的描述。
[0549]
表15示出了编码cascade i-e型的5种蛋白,具体地来自大肠杆菌菌株k-12mg1655的基因的多核苷酸dna序列,以及所得蛋白组分的氨基酸序列。基因组序列获自ncbi参考序列nz_cp014225.1。在表15中,多核苷酸序列是从大肠杆菌gdna扩增而来,或者是制造商生产的编码cascade蛋白组分的具体地针对在大肠杆菌中表达以及针对在人类细胞中表达进行了密码子优化的多核苷酸。
[0550][0551][0552]
另外,设计了包含cascade蛋白的几种融合蛋白。表16示出了编码cascade蛋白融合蛋白的基因的多核苷酸dna序列,以及所得蛋白组分的氨基酸序列。在大多数情况下,表16中描述的融合蛋白包括连接融合构建体中的两个多肽序列的较短的三氨基酸连接子;该连接子通常包含甘氨酸-甘氨酸-丝氨酸(ggs)或甘氨酸-丝氨酸-甘氨酸(gsg)。用于每种特定融合蛋白中的准确三氨基酸连接子序列可以在表16中的全长氨基酸序列中找到。
[0553][0554][0555]
当与其他cascade蛋白共表达时,cse2蛋白上的his6(六聚组氨酸;seq id no:418)和strep-tag
tm
ii(ge healthcare bio-sciences,pittsburgh,pa)(seq id no:419)肽标签使得能够分别经由镍-氮川乙酸(ni-nta)树脂或strep-tactin
tm
(iba gmbh llc,germany)树脂来纯化复合体。hrv3c(人类鼻病毒3c)蛋白酶识别序列(seq id no:420)被hrv3c蛋白酶切割,并且可用于从目标蛋白中去除n端融合物。cas6、cas7和/或cas8蛋白上的nls(核定位信号;seq id no:421肽标签使得能够在真核系统中进行核运输。
cas6或cas7蛋白上的ha(血凝素;seq id no:422)肽标签使得能够通过利用抗ha抗体的western印迹检测异源蛋白表达。mbp(麦芽糖结合蛋白;seq id no:423)肽融合是一种有利于纯化cas8蛋白的增溶标签。tev(烟草蚀纹病毒)蛋白酶识别序列(seq id no:424)被tev蛋白酶切割,并且可用于从目标蛋白中去除n端融合物。foki核酸酶域包含guo,et al.(guo,j.,et al.,j.mol.biol.400:96-107(2010))描述的sharkey变体,两个单体foki亚基联合形成同型二聚体,并在均二聚化后催化双链dna切割。连接子序列(seq id no:425)被用于融合foki核酸酶域至cas8蛋白。
[0556]
已经设计了不同长度和氨基酸组合物的另外的连接子序列,其将foki核酸酶域连接至cas8蛋白。这些氨基酸序列可以在表17中找到。
[0557][0558]
表18包含四个最小crispr阵列的多核苷酸dna序列,当转录成前体crrna并由cascade的rna核酸内切酶蛋白加工时,它们会生成在生化测定和细胞培养物基因编辑实验中用作靶向互补dna序列的向导rna的成熟的crrnas。
[0559]
最小的crispr阵列包含两个重复序列(带下划线的,小写字母),位于间隔区序列的两侧,其代表crrna的向导部分。由cascade核酸内切酶蛋白进行rna加工会在5'和3'端产生具有重复序列的crrna,位于向导序列的两侧。crispr阵列也可以通过由核酸内切酶cascade蛋白进行的rna加工扩展为包括位于两个间隔区序列两侧的三个重复序列(带下划线),其代表两个不同crrnas的向导部分。如果需要,可以进一步扩展阵列以包括另外的间隔区序列。
[0560]
[0561][0562]
实施例2
[0563]
用于产生cascade效应子复合体的细菌表达载体的设计
[0564]
本实施例描述了编码cascade相关蛋白,以及包含如实施例1所述的向导序列的最小crispr阵列的细菌表达载体的设计。描述了与编码最小crispr阵列的质粒一起使用的cascade亚基蛋白表达系统的构建。
[0565]
构建了单质粒cascade蛋白表达系统,以在大肠杆菌中表达称为casbcde复合体(其包含cse2、cas7、cas5和cas6蛋白,但不包含cas8蛋白)的cascade复合体的蛋白质,或在大肠杆菌中表达整个cascade cascade复合体的蛋白质。单个质粒系统在单个表达质粒上包含cse2
–
cas7
–
cas5
–
cas6操纵子,或整个cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子。cas8蛋白可以从其自身的表达质粒表达,用于生化实验,其中将其与casbcde复合体混合在一起以重构cascade。
[0566]
使用了用于表达载体构建的起始质粒(参见brouns,s.,et al.,science 321:960
–
964(2008))。包含cas操纵子的单质粒cascade蛋白表达系统按如下组装。将cas基因的编码序列按顺序cse2
–
cas7
–
cas5
–
cas6(casbcde复合体或cas8
–
cse2
–
cas7
–
cas5
–
cas6(完整cascade复合体)排列,并通过野生型细菌基因布置对应的序列(参见ncbi参考序列nz_cp014225.1)分离。
[0567]
为了附接编码亲和标签(his6或ii,iba gmbh llc,germany)的多核苷酸序列,将相应的编码序列插入cas8基因的3'端和cse2基因的5'端的连接处;这两个开放阅读框在野生型gdna序列中重叠。
[0568]
为了将编码n端nls和/或nls-ha标签的多核苷酸序列附接到cas6基因的5'端,在cas6和上游cas5基因之间引入了另外的间隔,因为这些开放阅读框在野生型gdna序列中重叠,使得cas6基因的shine-dalgarno序列位于cas5基因的3'部分中。在新的nls-cas6或nls-ha-cas6开放阅读框的上游插入了一个新的shine-dalgarno序列,以提高翻译效率。
[0569]
为了将编码c端nls和/或ha-nls标签的多核苷酸序列附接到cas7基因的3'端,在cas7和下游cas5基因之间引入了另外的间隔,因为这些开放阅读框在野生型gdna序列中非常接近,使得cas5基因的shine-dalgarno序列位于cas7基因的3'部分中。在新的cas7-nls或cas7-ha-nls开放阅读框的下游插入了一个新的shine-dalgarno序列,以提高cas5基因的翻译效率。
[0570]
为了将编码n端nls-foki-连接子融合物的多核苷酸序列附接到cas8蛋白上,在cas8基因的5
’
端插入了相应的编码序列。
[0571]
将cse2
–
cas7
–
cas5
–
cas6和cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子克隆到pcdf(milliporesigma,hayward,ca)载体骨架中,由于aada基因的存在其赋予了壮观霉素抗性。操纵子的转录由t7启动子驱动,并受lac操纵子控制;该载体还编码laci阻抑剂。t7终止子被克隆到cse2
–
cas7
–
cas5
–
cas6或cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子的下游。载体包含cdf复制起点。
[0572]
为了表达cas8或foki-cas8融合蛋白,将cas8基因克隆到pet(milliporesigma,hayward,ca)家族载体骨架中,由于kanr基因的存在其赋予了卡那霉素抗性。操纵子的转录由t7启动子(pt7)驱动,并受lac操纵子(laco)的控制;该载体还编码laci阻抑剂(laci基因)。t7终止子被克隆到cas8基因的下游。该载体包含cole1复制起点。
[0573]
图23a、图23b、图23c、图23d和图23e示出了cas8、foki
–
cas8、cse2
–
cas7
–
cas5
–
cas6操纵子、cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子和foki-cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子的过表达载体的示意图。图23a、图23b、图23c、图23d和图23e中的名称在本实施例(以及实施例1)中进行了描述,且如下所示:pt7(t7启动子)、laco(lac操纵子)、his6(六组氨酸)、mbp(麦芽糖结合蛋白)、ii(iba gmbh llc,germany)hrv3c(人类鼻病毒3c)蛋白酶识别序列、tev(烟草蚀纹病毒)蛋白酶识别序列、nls(核定位信号)、kanr(卡那霉素抗性基因)、laci(laci阻抑剂基因)、cole1 ori(复制起点)、cdf ori(clodf13复制起点)、foki核酸酶域(sharkey变体)和aada(编码氨基糖苷抗性蛋白的基因)。
[0574]
表19提供了编码cas8蛋白、casbcde复合体的4种蛋白(cse2
–
cas7
–
cas5
–
cas6操纵子)和cascade复合体的所有5种蛋白(cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子)的细菌表达质粒的序列。多核苷酸序列在cas8蛋白上提供有或没有n端foki融合。
[0575][0576]
为了纯化包含crrna的casbcde复合体和cascade复合体,将编码cse2
–
cas7
–
cas5
–
cas6操纵子或cas8
–
cse2
–
cas7
–
cas5
–
cas6操纵子的蛋白质表达载体与包含最小crispr阵
列的载体组合。
[0577]
crispr克隆被克隆到pacyc-duet1载体骨架中,该载体由于camr基因而赋予了氯霉素抗性。阵列的转录由t7启动子驱动,并受lac操纵子(laco)的控制;该载体还编码laci阻抑剂。t7终止子被克隆到crispr阵列的下游。该载体包含p15a复制起点。
[0578]
图24包含表达载体的示意图,该表达载体包含具有2个重复(图24,“重复区”)和1个间隔区(图24,“间隔区”)的crispr阵列。如本文所述,可以扩展该阵列。图24中的名称在本实施例(以及实施例1)中有描述,且如下所示:pt7(t7启动子)、laco(lac操纵子)、laci(laci阻抑剂基因)、p15a ori(复制起点)和camr(氯霉素抗性基因)。
[0579]
表20提供了编码最小crispr阵列的实例的细菌表达质粒的序列。
[0580][0581]
实施例3
[0582]
用于在哺乳动物细胞中产生cascade效应子复合体的真核表达载体的设计
[0583]
本实施例描述了编码cascade相关蛋白,以及包含如实施例1所述的组分序列的最小crispr阵列的真核表达质粒载体的设计。
[0584]
a.表达每种cascade蛋白和最小crispr阵列的单独的质粒
[0585]
可以通过在由人类巨细胞病毒(cmv)即刻早期启动子/增强子驱动且编码由人u6启动子驱动的单独的表达载体上的crrna的单独的表达载体上,编码每种蛋白组分来在哺乳动物细胞中表达cascade蛋白。
[0586]
每种表达质粒的起始质粒是pcdna3.1(thermo scientific,wilmington,de)的衍生物。针对在人类细胞中表达进行了密码子优化的(参见实施例1)cascade蛋白的编码序列,被插入到cmv启动子下游和牛生长激素(bgh)多腺苷酸化信号上游的载体中。将编码n端nls和3x-flag表位标签的cse2基因在5
’
端融合至多核苷酸序列。将编码n端nls的cas5基因在5
’
端融合至多核苷酸序列。将编码n端nls和ha表位标签的cas6基因在5
’
端融合至多核苷酸序列。将编码n端nls和myc表位标签的cas7基因在5
’
端融合至多核苷酸序列。将编码n端nls的cas8基因在5
’
端融合至多核苷酸序列;在另一个实施方案中,将编码n端nls、ha表位标签和foki核酸酶域的cas8基因在5
’
端融合至多核苷酸序列。
[0587]
将每个基因或基因融合物克隆到pcdna3.1衍生载体骨架中,其由于ampr基因的存在而赋予了氨苄青霉素抗性。由于neor基因的存在,该载体还编码新霉素抗性,它位于sv40早期启动子(psv40)和起点(sv40 ori)的下游,以及sv40早期聚腺苷酸化信号(sv40 pa)的上游。除了人cmv即刻早期启动子/增强子(pcmv)和bgh(牛生长激素)多聚腺苷酸化信号外,
载体还包含目标基因上游的t7启动子,允许进行mrna的体外转录。载体包含f1复制起点以及cole1复制起点。
[0588]
图25包含编码foki-cas8融合蛋白的哺乳动物表达载体的示意图。图25中的名称在本实施例(以及实施例1)中进行了描述,且如下所示:人cmv即刻早期启动子/增强子(pcmv)、nls(核定位信号)、foki(foki核酸酶域(sharkey变体))、cas8蛋白编码序列、bgh pa(牛生长激素一个聚腺苷酸信号)、f1 ori(f1噬菌体复制起点)、psv40(sv40早期启动子)、sv40 ori(sv40起点)、neor(新霉素抗性基因)、sv40 pa(sv40早期聚腺苷酸化信号)、cole1 ori(复制起点)和ampr(氨苄青霉素抗性基因)。类似地设计了编码其他cascade蛋白的载体。
[0589]
表21提供了编码cse2、cas5、cas6、cas7、cas8和foki-cas8中的每种的单个哺乳动物的表达载体的序列。
[0590][0591]
crisprrna用最小crispr序列编码,该序列包含位于两个间隔区序列两侧的三个重复区。可以利用最小阵列中的最外侧重复区两侧的另外的序列设计产生crisprrna的构建体。通过cascade复合体的rna加工蛋白(cas6蛋白)使得能够对前体crisprrna进行处理,该蛋白可以在单独的质粒上表达。
[0592]
crispr阵列被克隆到上述相同的pcdna3.1衍生载体骨架中,除了人cmv启动子被人u6启动子(pu6)取代,并且bgh聚腺苷酸化信号被多聚t终止信号取代之外。图35中示出了此类crispr阵列的实例。在图中,hu6启动子(图35,显示为点状区域)与第一重复序列(空白方块)相邻,第一重复序列与第一间隔区序列(图35,间隔区1,斜线)相邻,第一间隔区序列与第二重复序列(图35,灰色方形)相邻,第二重复序列与第二间隔区序列(图35,间隔区2)相邻,第二间隔区序列与第三重复序列(图35,黑色方形)相邻。在图35中,示出了包含成对的grna向导的区域(图35,成对的grnas)。
[0593]
图26包含编码靶向trac基因的代表性crispr阵列的真核表达载体的示意图。在本实施例(以及实施例1)中描述了图26中的名称,且如下所示:pu6(人u6启动子)、重复区(crisprrna重复区)、trac间隔区-1(靶向trac基因的第一间隔区)、trac间隔区-2(靶向trac基因的第二间隔区)、多聚t(多聚t终止信号)、f1 ori(f1噬菌体复制起点)、psv40(sv40早期启动子)、sv40 ori(sv40起点)、neor(新霉素抗性基因)、sv40 pa(sv40早期聚腺苷酸信号)、cole1 ori(复制起点)和ampr(氨苄青霉素抗性基因)。
[0594]
表22提供了代表性哺乳动物的编码靶向trac基因的crispr阵列的表达载体的序列;靶向trac基因中的匹配性dna序列的间隔区序列可以在表18中找到。
[0595][0596]
b.其中从单个启动子表达多种cascade蛋白编码序列的cascade蛋白表达系统
[0597]
为了从更少的表达载体表达cascade复合体的组分,构建了多顺反子的表达载体。在每一个上,单个cmv启动子同时驱动多个编码序列的表达,这些编码序列被2a病毒肽序列隔开。一点褐翅蛾(thosea asigna)病毒2a肽序列诱导核糖体跳跃(参见,例如liu,z.,et al.,sci.rep.7:2193(2017)),从而使多个蛋白质编码基因能够串联在单个多顺反子的构建体中。
[0598]
多顺反子的表达质粒的起始质粒是上述pcdna3.1相同的衍生物,包含cmv启动子和bgh聚腺苷酸化信号。将针对在人类细胞中表达进行了密码子优化的(参见实施例1)的cascade蛋白的编码序列以cas7-cse2-cas5-cas6-cas8的顺序连接,其中编码一点褐翅蛾病毒2a(t2a)肽的多核苷酸序列插入在每对基因之间。另外,在每个cascade蛋白基因的5'端附接了编码nls标签的多核苷酸序列,并且编码foki核酸酶域的多核苷酸序列被附接至cas8基因的5
’
端,通过30个氨基酸连接子序列连接。最终的构建体具有以下顺序的元件:nls-cas7-t2a-nls-cse2-t2a-nls-cas5-t2a-nls-cas6-t2a-nls-foki-连接子-cas8。
[0599]
图27包含编码所有cascade蛋白的示例性的多顺反子的哺乳动物表达载体的示意图。在本实施例(以及实施例1)中描述了图27中的名称,且如下所示:人cmv即刻早期启动子/增强子(pcmv)、nls(核定位信号)、t2a(编码一点褐翅蛾病毒2a肽的多核苷酸序列)、cas7、cse2、cas5和cas6蛋白的编码序列、foki(foki核酸酶域(sharkey变体)连接子序列、cas8蛋白的编码序列、bgh pa(牛生长激素聚腺苷酸化信号)、f1 ori(f1噬菌体复制起点)、psv40(sv40早期启动子)、sv40 ori(sv40起点)、neor(新霉素抗性基因)、sv40 pa(sv40早期聚腺苷酸化信号)、cole1 ori(复制起点)、ampr(氨苄青霉素抗性基因)和mlui限制位点。
[0600]
表23提供了编码所有cascade蛋白的示例性的多顺反子的哺乳动物表达载体的序列。该载体可以与编码上述crisprrna的哺乳动物的表达载体联合,以在哺乳动物细胞中产生功能性的cascade复合体。
[0601][0602]
c.单质粒表达系统
[0603]
构建了单质粒cascade表达系统,以在人类细胞中表达完整的cascade复合体。质粒在单个质粒上编码整个cas8
–
cse2
–
cas7
–
cas5-cas6操纵子和最小crispr阵列。通过将最
小crispr阵列连同上游人u6启动子和下游多聚t终止信号一起插入mlui限制性位点,从多顺反子的蛋白表达载体(如表23和图27中所述)构建该质粒。
[0604]
表24提供了用于表达所有5种cascade蛋白以及crrna以促进在人类细胞中形成cascade复合体的单质粒的序列。
[0605][0606]
还设计了用于在大肠杆菌和哺乳动物细胞中表达cas3蛋白(seq id no:21;单体cas3核酸酶/解旋酶大肠杆菌k-12亚菌株mg1655)的质粒。
[0607]
表25提供了这些质粒的构建体和序列。
[0608][0609]
实施例4
[0610]
将编码cascade组分的多核苷酸引入细菌生产菌株中
[0611]
本实施例描述了使用大肠杆菌表达系统在细菌细胞中引入和表达cas8亚基蛋白编码序列,以及改造的i型crispr-cas效应子复合体的组分的编码序列。
[0612]
a.cas8蛋白的表达
[0613]
从包含用于来自t7启动子的his6-mbp-tev-cas8的iptg可诱导的表达的操纵子的质粒(实施例2,seq id no:438,表19,图23a)表达大肠杆菌i-e型cas8蛋白。表达质粒赋予了对卡那霉素的抗性。
[0614]
为了表达cas8蛋白,用表达质粒转化了大肠杆菌细胞。简言之,将微量离心管中的100μl等分的化学感受态的大肠杆菌细胞(大肠杆菌bl21 startm(thermo fisher scientific,waltham,ma)细胞)在冰上融化10分钟。将35ng的质粒dna添加至解冻的细胞中,并将细胞与dna一起在冰上孵育8分钟。通过将微量离心管置于42℃水浴中30秒,然后立即将管置于冰中2分钟来进行热激。将900μl的2xyt培养基添加至微量离心管中,并将微量
离心管置于37℃的管旋转器中1小时。最后,将100μl的回收细胞接种在lb固体卡那霉素(50μg/ml)上,并在37℃下孵育过夜。
[0615]
从在抗生素选择板上生长的集落中选出单个菌落,并接种到10ml补充有卡那霉素(50μg/ml)的2xyt培养基中。将培养物在37℃下培养过夜,同时在定轨振荡器中以200rpms的速度振荡。将6ml过夜培养物转移至装有1l补充有卡那霉素(50μg/ml)的2xyt培养基的2l带挡板的烧瓶中。将1l培养物在37℃下培养,同时在定轨振荡器上以200rpm的速度振荡,直到600nm下的光密度为0.56。
[0616]
然后通过添加iptg至终浓度为1mm来诱导表达。将诱导的培养物在16℃下培养过夜,同时在定轨振荡器中以200rpm的速度振荡。通过在4℃下以4,000rcf离心15分钟来收获细胞。将细胞沉淀重悬于15ml裂解缓冲液中,该裂解缓冲液由50mm tris ph 7.5、100mm nacl、5%甘油和1mm tcep组成,其中每50ml裂解缓冲液中补充有1片completetm(roche,basel,switzerland)蛋白酶抑制剂片。将重悬的细胞转移至50ml锥形管中,以立即进行下游处理。纯化cas8蛋白,并基本上按以下针对foki-foki-cas8融合蛋白所述(实施例5c)对纯化的蛋白进行定征。
[0617]
b.cascade rnp复合体组分的表达
[0618]
使用双质粒系统,在大肠杆菌细胞中共表达5种大肠杆菌cascade蛋白和rna向导的完整集合,以产生cascade rnp复合体。一个质粒(实施例2,seq id no:441,表19,图23d)包含来自t7启动子的用于cse2、cas5、cas6、cas7和cas8蛋白的iptg可诱导的表达的操纵子。包括his6亲和标签作为与cse2的n端的翻译融合(实施例1,seq id no:392,表16)。第二质粒编码iptg可诱导表达的j3向导(实施例2,seq id no:444,表20,图24)。cascade蛋白表达质粒赋予了壮观霉素抗性,并且cascade rna向导表达质粒赋予了氯霉素抗性。
[0619]
为了在同一细胞中共表达cascade蛋白和rna组分,同时用两种质粒转化了大肠杆菌细胞。将于微型离心管中的100μl等分的化学感受态大肠杆菌细胞(大肠杆菌,bl21 startm(de3)(thermo fisher scientific,waltham,ma))在冰上解冻10分钟。将35ng的每种质粒加入解冻的细胞中,并将细胞与dna一起在冰上孵育8分钟。通过将微量离心管置于42℃的水浴中30秒,然后立即将微量离心管置于冰中2分钟来进行热激。将900μl的2xyt培养基添加到微量离心管中,并将微量离心管置于37℃的管旋转器中1小时。最后,将100μl回收的细胞接种到含有氯霉素(34μg/ml)和壮观霉素(50μg/ml)的lb固体培养基上,并在37℃下孵育过夜。
[0620]
从生长在抗生素选择板上的集落中选出单个集落,并接种到10ml补充了氯霉素(34μg/ml)和壮观霉素(100μg/ml)的2xyt培养基中。将培养物在37℃下培养过夜,同时在定轨振荡器中以200rpms的速度振荡。将6ml过夜培养物转移至装有补充了氯霉素(34μg/ml)和壮观霉素(100μg/ml)的1l 2xyt培养基的2l带挡板的烧瓶中。将1l培养物在37℃下培养,同时在轨道振荡器中以200rpm的速度振荡,直到在600nm处的光密度为0.56。
[0621]
通过添加iptg至终浓度1mm来诱导两种质粒的表达。将诱导的培养物在16℃下培养过夜,同时在定轨振荡器中以200rpm的速度振荡。通过在4℃下以4,000rcf离心15分钟来收获细胞。将细胞沉淀重悬于15ml裂解缓冲液中,该裂解缓冲液由50mm tris ph 7.5、100mm nacl、5%甘油和1mm tcep组成,其中每50ml的裂解缓冲液中补充了1片completetm(roche,basel,switzerland)蛋白酶抑制剂片。将重悬的细胞转移至50ml锥形管中,以立即
进行下游处理。按如下所述纯化和定征cascade rnp复合体。
[0622]
实施例5
[0623]
cascade组分和cascade rnp复合体的纯化
[0624]
本实施例描述了纯化通过按实施例4b中所述在细菌中过表达产生的大肠杆菌i-e型cascade rnp复合体的方法。该方法使用固定的金属亲和色谱,然后进行尺寸排阻色谱(sec)。本实施例还描述了用于评估纯化的cascade rnp产品质量的方法。另外,本实施例描述了cascade组分的纯化和定征。
[0625]
a.cas8、cas7、cas6、cas5和cse2 cascade rnp复合体的纯化
[0626]
按实施例4b中所述产生大肠杆菌i-e型cascade rnp复合体。使用固定的金属亲和色谱法捕获cascade复合体。简言之,将按实施例4b所述产生的重悬细胞沉淀在冰上解冻,并通过另外15ml的裂解缓冲液来使体积达到35ml,该裂解缓冲液由50mm tris ph 7.5、100mm nacl、5%甘油和1mm tcep组成,其中每50ml裂解缓冲液中补充有1片completetm(roche,basel,switzerland)蛋白酶抑制剂片。
[0627]
将50ml锥形管置于冰水浴中,并使用带有1/2英寸尖端的q500超声仪(qsonica,newtown,ct)通过两轮超声处理裂解细胞。每一轮超声处理由2.5分钟的处理周期组成,重复周期为50%振幅下超声处理10秒,然后休息20秒。在几轮超声处理之间,允许试管在冰水浴中冷却1分钟。通过在4℃下以48,384rcf离心30分钟来澄清裂解物。然后将澄清的上清液添加到hispurtm ni-nta(thermo fisher scientific,waltham,ma)树脂中,该树脂已用由50mm tris ph 7.5、100mm nacl、10mm咪唑、5%甘油和1mm tcep组成的ni洗涤缓冲液预平衡。每1l大肠杆菌表达培养物使用1.5ml床体积的镍亲和树脂。在轻微混合下于4℃孵育1小时后,通过在4℃下以500rcf离心2分钟来沉淀树脂。吸出上清液,并用5倍床体积的ni洗涤缓冲液洗涤树脂5次。每次洗涤后,将树脂在4℃下于500rcf下沉淀2分钟,并通过抽吸除去上清液。最后,通过添加五倍床体积的ni洗脱缓冲液洗脱结合的蛋白质(包括cascade rnp复合体),ni缓冲液由50mm tris ph 7.5、100mm nacl、300mm咪唑、5%甘油和1mm三(2-羧乙基)膦(tcep)组成。在4℃下以500rcf离心2分钟后,将镍亲和洗脱液吸入干净的50ml锥形管中。
[0628]
将镍亲和洗脱液通过尺寸排阻色谱法(sec)进一步纯化。使用具有-50(milliporesigma,hayward,ca)膜的(milliporesigma,billerica,ma)超滤旋转浓缩器,在12℃下通过超滤将镍亲和洗脱液浓缩至0.5ml的最终体积。使用0.22μm ultrafree-mc gv(milliporesigma,hayward,ca)离心过滤器过滤浓缩的样品,然后通过在用由50mm tris ph 7.5、500mm nacl、5%甘油、0.1mm edta和1mm tcep的sec缓冲液平衡的hipreptm 16/60s-300(ge healthcare,uppsala,sweden)柱上以0.5ml/分钟的流速于4℃下分离来进一步纯化。用sec缓冲液洗脱蛋白质,并收集1ml馏分。如通过uv 280判断的,最早的洗脱峰被认为是高分子量的聚集物质,并且相应的馏分被丢弃。通过考马斯染色的sds-page分析随后的洗脱馏分。每种适当形成的复合体均包含一个分子的cas8、六个分子的cas7、一个分子的cas6和cas5中每种,以及两个分子的cse2。合并在sds-page凝胶上观察时具有近似cascade蛋白的预期化学计量的洗脱馏分。通过分光光度法对合并的馏分进行分析,以确认它们包含大量的核酸组分,如通过在260nm处的吸光度大于在280nm处的吸光度所证明的。
[0629]
通过用具有(milliporesigma,hayward,ca)膜的(milliporesigma,hayward,ca)旋转浓缩器将合并的样品浓缩至100ul然后用储存缓冲液稀释50倍,将合并的样品交换至由50mm tris ph 7.5、100mm nacl、5%甘油、0.1mm edta和1mm tcep组成的存储缓冲液中。最后,使用相同的超滤装置将样品浓缩至10mg/ml,并保存在-80℃下。
[0630]
用分光光度法分析最终纯化的产物,以确定cascade rnp复合体的最终浓度,并确认存在核酸组分,如通过260nm处的吸光度大于280nm处的吸光度所证明的。通过将280nm处的吸光度除以路径长度为1cm的完整复合体的0.1%溶液的计算吸光度来确定cascade rnp复合体的浓度。纯化的复合体的0.1%溶液的预测吸光度为2.03cm-1
,并通过将复合体中每种分子在280nm下的计算消光系数(916940m-1
cm-1
)的总和除以复合体中每种分子的分子量的总和(450832g/mole)来计算。
[0631]
另外,通过sds-page用考马斯亮蓝染色分析最终产物以确认每种蛋白质组分均以大约正确的化学计量存在,并评估了污染蛋白的存在。将sds-page凝胶用考马斯instantbluetm(expedeon,san diego,ca)染色剂染色。使用gel doctm ez(bio-rad,hercules,ca)成像仪对凝胶成像,并使用imagelab(bio-rad,hercules,ca)软件注释。
[0632]
b.包含cas7、cas6、cas5和cse2蛋白的cascade复合体的纯化
[0633]
纯化了由蛋白组分cas7、cas6、cas5和cse2构成的cascade复合体。基本上按实施例4b中所述从第一质粒(实施例2,图24)表达l3向导rna(实施例2,seq id no:445,表20)。基本上按实施例4b中所述从第二质粒(实施例2,seq id no:440,表19、图23c)表达cascade蛋白。
[0634]
使用亲和色谱法捕获复合体。将重悬的细胞沉淀在冰上解冻。在50ml锥形管中,用另外的15ml裂解缓冲液调节体积升至35ml,该裂解缓冲液由50mm tris ph 7.5、100mm nacl、5%甘油、1mm tcep组成,其中每50ml裂解缓冲液中补充有1片completetm(roche,basel,switzerland)蛋白酶抑制剂片。将50ml锥形管置于冰水浴中,并使用具有1/2英寸尖端的q500超声仪(qsonica,newtown,ct)通过六轮超声处理裂解细胞。每一轮超声处理由一个1分钟的处理周期和重复循环的90%振幅下3轮超声处理然后休息9秒组成。在数轮超声处理之间,允许试管在冰水浴中冷却一分钟。通过在4℃下以48,384rcf离心30分钟来澄清裂解液。将澄清的上清液通过加入(iba gmbh llc,germany)树脂来进行亲和纯化,该树脂已经用由50mm tris ph 7.5、100mm nacl、1mm edta、5%甘油和1mm tcep组成的strep-wash缓冲液预平衡。每1l大肠杆菌表达培养物使用0.55ml床体积的亲和树脂。在4℃轻微混合下孵育一小时后,将样品倒入30ml一次性重力流柱(bio-rad,hercules,ca)上,允许未结合的物质流过柱子。用五倍床体积的strep洗涤缓冲液洗涤树脂五次。最后,将结合的蛋白用两次连续添加的五倍床体积的strep洗脱缓冲液洗脱,该缓冲液由50mm tris ph 7.5、100mm nacl、2.5mm脱硫生物素、5%甘油、1mm edta和1mm tcep组成。
[0635]
将亲和洗脱液通过sec进一步纯化。通过使用具有(milliporesigma,hayward,ca)膜的(milliporesigma,hayward,ca)旋转浓缩器在12℃下超滤,将亲和洗脱液浓缩至550ul的最终体积。使用0.22μm 13mm
(santa cruz biotechnology,dallas,tx)pvdf注射器过滤器过滤浓缩的样品,然后通过在4℃下以0.4ml/分钟的流速在用由50mm tris ph 7.5、500mm nacl、5%甘油、0.1mm edta和1mm tcep组成的sec缓冲液平衡的hipreptm 16/60s-300(ge healthcare,uppsala,sweden)柱上分离来进一步纯化。用sec缓冲液洗脱蛋白,并收集0.75ml馏分。如通过uv 280判断的,最早的洗脱峰被认为是高分子量的聚集物质,并且相应的馏分被丢弃。将与第二峰对应的馏分(第一个uv 280峰背面上的一个肩峰)合并。
[0636]
通过用具有(milliporesigma,hayward,ca)膜的(milliporesigma,hayward,ca)旋转浓缩器浓缩低至200ul然后用存储缓冲液稀释75倍,来将合并的样品交换至由50mm tris ph 7.5、100mm nacl、5%甘油、0.1mm edta和1mm tcep组成的存储缓冲液中。将样品第二次浓缩至700ul,然后再次用存储缓冲液稀释20倍。最后,在相同的超滤装置中将样品浓缩至4.7mg/ml,并保存在-80℃下。
[0637]
用分光光度法分析最终纯化的产物,以确定cascade rnp复合体的最终浓度,并确认存在核酸组分,如通过260nm处的吸光度大于280nm处的吸光度所证明的。通过将280nm处的吸光度除以路径长度为1cm的完整复合体的0.1%溶液的计算吸光度来确定cascade rnp复合体的浓度。纯化后的复合体的0.1%溶液的预测吸光度为2.18cm-1
,并且通过将复合体中每种分子在280nm处的计算消光系数的总和(762240m-1
cm-1
)除以复合体中每种分子的分子量的总和(348952.07g/摩尔)来进行计算。
[0638]
另外,通过sds-page用考马斯亮兰染色分析最终产物,以确认每种cascade蛋白均以大约正确的化学计量存在,并评估污染蛋白的存在。将sds-page凝胶用考马斯instantbluetm(expedeon,san diego,ca)染色剂染色。使用gel doctm ez(bio-rad,hercules,ca)成像仪对凝胶成像,并使用imagelab(bio-rad,hercules,ca)软件注释。每种适当形成的复合体均包含六个分子的cas7、一个分子的cas6和cas5中的每种,以及两个分子的cse2。
[0639]
c.foki-cas8融合蛋白的纯化
[0640]
本文中描述了使用固定的金属亲和色谱、阳离子交换层析(ciex)和最终的尺寸排阻色谱(sec),从细菌过表达的沉淀物中纯化包含融合至大肠杆菌i-e型cas8蛋白的foki核酸酶的融合蛋白的方法。
[0641]
在实施例1中描述了包括连接子序列的大肠杆菌i-e型foki-cas8融合蛋白(seq id no:413,表16)。在实施例2中描述了表达质粒(seq id no:439,表19,图23b)。基本上按实施例4a中所述产生包含融合蛋白的细胞。cas8融合蛋白包含n端his6标签、麦芽糖结合蛋白域、tev切割位点、foki核酸酶域和30个氨基酸的连接子。使用固定的金属亲和色谱法捕获蛋白质。将含有重悬细胞团的50ml圆锥管在冰上解冻。然后将试管置于冰水浴中,并使用具有1/4英寸尖端的q500超声仪(qsonica,newtown,ct),通过以重复循环的以40%振幅下超声处理10秒然后休息20秒,超声处理三分钟的处理周期来裂解细胞。通过在4℃以30,970rcf离心30分钟使裂解液澄清。然后将澄清的上清液添加到hispurtm ni-nta(thermo fisher scientific,waltham,ma)树脂中,该树脂已用由50mm tris ph 7.5、100mm nacl、10mm咪唑、5%甘油和1mm tcep组成的ni洗涤缓冲液预平衡。将2ml床体积的镍亲和树脂用于1l大肠杆菌表达培养物。在4℃下于轻微混合下孵育一小时后,将样品倒入30ml一次性重力流柱(bio-rad,hercules,ca)上,允许未结合的物质流过色柱子。用五倍床体积的ni洗涤
缓冲液洗涤树脂五次。最后,用五倍床体积的ni洗脱缓冲液洗脱结合的蛋白质,该缓冲液由50mm tris ph 7.5、100mm nacl、300mm咪唑、5%甘油和1mm tcep组成。
[0642]
用tev蛋白酶处理镍亲和洗脱液以除去亲和标签。将tev蛋白酶以1:25(w/w)的比例加入洗脱液中。使用12ml slid-a-lyzertm、10k mwco(thermo fisher scientific,waltham,ma)透析盒,将包括tev在内的样品对ni洗涤缓冲液透析过夜。
[0643]
通过ni亲和色谱从透析样品中除去tev蛋白酶和裂解的his6-mbp片段。将透析的样品倒至用镍洗涤缓冲液平衡过的干净的hispurtm ni-nta(thermo fisher scientific,waltham,ma)树脂柱上。然后用1倍柱体积的ni-nta洗涤缓冲液洗涤树脂。使用具有(milliporesigma,hayward,ca)膜的(milliporesigma,hayward,ca)旋转浓缩器,将流过液和洗涤液合并、浓缩,并交换到存储缓冲液(50mm tris ph 7.5、500mm nacl、5%甘油和1mm tcep)中。然后将该样品在-80c下冷冻保存。
[0644]
将样品解冻,并通过阳离子交换色谱法(ciex)进一步纯化。将样品在冰上解冻,并用由50mm tris ph 7.5、5%甘油和1mm tcep组成的冷的ciex_a缓冲液将其稀释10倍,从0.475ml稀释至4.75ml,导致50mm nacl的最终浓度。使用10ml毛细环将样品加载到用包含ciex_a缓冲液和5%ciex_b缓冲液(50mm tris ph 7.5、1m nacl、5%甘油和1mm tcep)的缓冲液平衡的1ml hitraptm sp hp(ge healthcare,uppsala,sweden)柱上。整个分离过程中的流速为0.75ml/min。用15ml的5%ciex_b缓冲液将环倒空到柱上。用另外的2ml 5%ciex_b缓冲液洗去未结合的样品。在将结合的蛋白用8ml线性梯度从5%至65%的ciex_b缓冲液洗脱时,收集500μl馏分。有两个主要的uv280洗脱峰。合并了对应于这两个峰中的第一个的四种馏分。总合并体积为2ml。
[0645]
将合并的ciex馏分通过sec进一步纯化。通过使用具有(milliporesigma,hayward,ca)膜的(milliporesigma,hayward,ca)旋转浓缩器,在12℃通过超滤将合并的ciex馏分浓缩至0.3ml的最终体积。使用0.22μm ultrafree-mc gv离心(milliporesigma,hayward,ca)旋转过滤器过滤浓缩的样品(,并通过在4℃下以0.6ml/分钟的流速在用cas8 sec缓冲液(50mm tris ph 7.5、200mm nacl、5%甘油和1mm tcep平衡过的10/300superdextm 200gl increase(ge healthcare,uppsala,sweden)柱上分离进一步纯化)。用cas8 sec缓冲液洗脱蛋白,并收集0.5ml馏分。如通过uv 280判断的,最早的洗脱峰被认为是高分子量的聚集物质,并且相应的馏分被丢弃。约14ml后洗脱出第二主要的uv 280峰。合并了与此第二峰相对应的馏分。将合并的样品用具有(milliporesigma,hayward,ca)膜的(milliporesigma,hayward,ca)旋转浓缩器浓缩至40μl。将浓缩的样品储存在-80℃下。
[0646]
用分光光度法分析最终纯化的产物,以确定融合蛋白的最终浓度,并确认不存在大量的核酸组分,如通过在280nm处的吸光度大于在260nm处的吸光度所证明的。通过将280nm处的吸光度除以完整复合体的0.1%溶液的计算吸光度来确定foki-cas8融合物的浓度。纯化的复合体的0.1%溶液的预测吸光度为1.05cm-1
,并通过将foki-cas8融合物在280nm处的消光系数(86290m-1
cm-1
)除以其分子量(82171.32g/摩尔)来计算。另外,通过用instantbluetm(expedeon,san diego,ca)染色剂染色的sds-page凝胶分析最终产物。使用gel doctm ez(bio-rad,hercules,ca)成像仪对凝胶成像,并使用imagelab(bio-rad,
hercules,ca)软件注释。该分析表明,纯化的融合蛋白具有预期的大小,并且仅存在低水平的污染蛋白。
[0647]
实施例6
[0648]
用于生化切割测定的dsdna靶序列的产生
[0649]
用于cascade或cascade-融合效应子复合体的体外dna结合或切割测定的dsdna靶序列可以使用几种不同的方法产生。本实施例描述了三种产生靶序列的方法,包括合成ssdna寡核苷酸的退火、从gdna中经pcr扩增选择的核酸靶序列,和/或将核酸靶序列克隆到细菌质粒中。将dsdna靶序列用于cascade结合或切割测定。
[0650]
a.通过退火合成的ssdna寡核苷酸来产生dsdna靶序列
[0651]
从商业制造商(integrated dna technologies,coralville,ia)购买了编码包含通过crisprrna的向导部分识别的靶序列、临近的前间区序列邻近基序(pam)和另外的5
’
和3
’
两侧序列的目标靶区的dna寡核苷酸。每个构建体订购了两种寡核苷酸,一种包含有义链,且一种包含无义链。
[0652]
表26列出了被订购为包含来源于噬菌体λgdna的表示为j3的靶序列的寡核苷酸序列。在5'和3'端,靶标和pam序列两旁有20bp另外的序列。
[0653][0654]
通过将等摩尔浓度(10μm)的两种寡核苷酸在1x退火缓冲液(6mm hepes,ph 7.0和60mm kcl)中混合,在95℃下加热2分钟,然后缓慢冷却来将寡核苷酸退火。然后将退火的寡核苷酸与cascade和/或cascade-效应子域融合rnps一起直接用于dna结合和/或dna切割测定中。
[0655]
编码包含crisprrna的向导部分识别的靶序列以及两侧的临近的前间区序列邻近基序(pam),以及另外的5
’
和3
’
两侧序列的目标靶区的5
’
cy5荧光标记的dna寡核苷酸购自商业制造商(integrated dna technologies,coralville,ia)。每种构建体订购了四种寡核苷酸,一种包含5'荧光标记的有义链,一种包含5'未标记的有义链,一种包含5'荧光标记的无义链,且一种包含5'未标记的无义链。在5'和3'端,靶标和pam序列两侧有20bp另外的序列。
[0656]
表27列出了订购为包含来源于噬菌体λgdna的表示为j3的靶序列和来源于人ccr5位点的表示为ccr5的对照靶序列的寡核苷酸序列。
[0657][0658]
通过在1x退火缓冲液(6mm hepes,ph 7.0,60mm kcl)中以等摩尔浓度(1μm)混合标记和未标记的或两种标记的或两种未标记的寡核苷酸,在95℃下加热2分钟,然后缓慢冷却,来将寡核苷酸退火。然后将退火的寡核苷酸与cascade和/或cascade-效应子域融合rnps一起直接用于dna结合测定中。将cy5荧光标记的dna寡核苷酸用azure c600(azure biosystems,dublin,ca)生物成像仪成像。
[0659]
本方法可用于产生另外的标记或未标记的靶标或双靶标序列,从而双靶标定义为包含被通过间隔区间序列隔开的单独的cascade分子靶向的两个前间隔区序列的靶标。
[0660]
b.通过从gdna pcr扩增来产生dsdna靶序列
[0661]
使用pcr扩增直接从gdna模板物质产生来源于人gdna的双靶标的dsdna靶序列。具体来说,pcr反应包含从k562细胞纯化的人gdna和q5热启动高保真2x master mix(new england biolabs,ipswich,ma),以及表28中列出的引物,其中带下划线的部分对应于gdna中的引物结合位点。
[0662][0663]
根据制造商的说明书(new england biolabs,ipswich,ma)进行pcr,并使用nucleospin凝胶和pcr清洁试剂盒(macherey-nagel,bethlehem,pa)纯化长度为288bp的所需产物dna。然后将该dsdna与cascade和/或cascade-效应子域融合rnps一起直接用于的dna结合和/或dna切割测定中。
[0664]
c.通过将靶序列克隆至细菌质粒中来产生dsdna靶序列
[0665]
编码包含被crisprrna的向导部分识别的靶序列、临近的前间区序列邻近基序(pam)和另外的5
’
和3
’
两侧序列的也称为前间隔区的目标靶区的dna寡核苷酸购自商业制造商(integrated dna technologies,coralville,ia)。设计了寡核苷酸,使得当退火时,在通过限制酶ecori和blpi或通过bamhi和ecori切割其各自的识别位点后,末端再生粘性末端。寡核苷酸被设计为包含来源于噬菌体λ基因组的表示为j3的单个靶序列。另外,寡核苷酸被设计成包含来源于噬菌体λ基因组的通过15bp间隔区间序列彼此分开的表示为j3和l3的两个串联靶序列。这些寡核苷酸的序列在表29中列出。
[0666][0667]
寡核苷酸含有5'磷酸化末端,其由商业制造商引入或在内部使用t4多核苷酸激酶(new england biolabs,ipswich,ma)进行磷酸化。然后,通过将等摩尔量的退火缓冲液(6mm hepes,ph 7.0,60mm kcl)混合在一起,加热至95℃ 2分钟,然后在台上缓慢冷却,以1μm的最终浓度将寡核苷酸退火。
[0668]
单独地,将pacyc-duet1(milliporesigma,hayward,ca)质粒用相应的一对限制性酶bamhi和ecori或ecori和blpi进行双消化,其粘性末端与由杂交的寡核苷酸的末端形成的粘性末端匹配。使用琼脂糖凝胶电泳将双消化的载体与去除的插入物分离。
[0669]
为了将杂交的寡核苷酸克隆到双消化的载体中,将杂交的寡核苷酸稀释至50nm的储备浓度,然后使用杂交的寡核苷酸、双消化的载体和和快速连接酶(new england biolabs,ipswich,ma)形成10μl的连接反应物。然后将连接反应物用于转化化学感受态的大肠杆菌菌株,并在琼脂糖平板上培养过夜后,将单个克隆分离并在液体培养物中培养以产生足够的细菌培养物,以从中分离质粒。然后使用sanger测序来验证所需的质粒序列。表30提供了包含j3靶序列的质粒(seq id no:481)和包含通过15bp间隔区间序列隔开的j3和l3靶标序列的质粒(seq id no:482)的完整载体序列。
[0670][0671]
其他的克隆操作被用于产生另外的双靶标质粒构建体。seq id no:482的15bp的间隔区间序列包含独特的avrii和xhoi限制位点。因此,将另外的杂交的寡核苷酸引入这些限制位点,可以将间隔区间扩展到更长的长度,以便用纯化的cascade和cascade-核酸酶融合rnps进行生化测试。因为crrna引导的foki-cascade融合复合体靶向两个相邻的dna位点,所以来自相邻的dna结合的复合体的foki域的二聚化导致在将两个靶标位点隔开的间隔区间内进行dna切割。设计并测试了可变的间隔区间长度,以评估在foki核酸酶域和其融合的cascade亚基蛋白之间具有给定束缚几何学的给定间隔区间长度。在表30中作为seq id no:483给出了包含30bp的扩展的间隔区间序列的靶标dna底物的完整载体序列。
[0672]
此外,以下克隆策略提供了包含沿一个大插入物串连连接的数个靶序列的质粒底物。从商业制造商(integrated dna technologies,coralville,ia)订购了基因块,其包含17个连续的双靶标。该基因块包含将每个双靶标与临近的双靶标隔开的4bp片段,并且包含来源于智人gdna的16个双靶标,以及包含来源于噬菌体γ基因组的j3/l3靶标的一个对照双靶标。表31中显示了16个连续的人双靶标的基因组坐标。该基因块被订购为在末端具有两侧的saci和sbfi限制位点,使得可以将其克隆到pacyc-duet1载体中的saci和sbfi位点中。通过将基因块克隆到pacyc-duet1中而产生的多靶标质粒底物的完整载体序列在表30中示出为seq id no:484。这种多靶标序列质粒允许对具有靶向质粒内的连续连接的靶标位点中的一个的crrnas的多种不同的foki-cascade制剂进行生化测试。
[0673][0674]
实施例7
[0675]
纯化的cascade复合体在生化切割测定中的应用
[0676]
本实施例示出了foki-cascade融合蛋白复合体在生化dsdna切割测定中的应用。根据其在dsdna切割中的活性来比较蛋白试剂。
[0677]
设计了来源于大肠杆菌i-e型cascade系统的foki
–
cascade rnps,在大肠杆菌中重组表达,并纯化后使用,如实施例1、2和5中所述。这些rnps被设计为包含靶向来源于噬菌体γgdna的j3和l3靶序列,或靶向人gdna中的trac基因中的内含子的crispr rnas。每种rnp制剂都是包含两种foki-cascade复合体的异质混合物,这些复合体在其他方面是相同的,除了crrna的向导部分外。
[0678]
分别从缺少cas8的(cas8-less)cascade复合体纯化foki-cas8,用靶向j3和l3γ靶序列的向导多核苷酸程序化,并以pam在内的配置用于利用具有靶标位点的j3/l3质粒底物的生化切割测定中。
[0679]
通过将casbcde复合体(使用seq id no:440和seq id no:446产生的,如实施例2中所述)与包含16-aa连接子的纯化的foki-cas8(在实施例2中描述了一般的foki-cas8表达载体序列,表19中的seq id no:439;特定的16-aa连接子在实施例1中,表17中的seq id no:431)混合在一起,重构了foki-cascade复合体。在1x cascade切割缓冲液(20mm tris-cl,ph 7.5,200mm nacl,5mm mgcl2,1mm tcep,5%甘油)中,利用1μm最终浓度的casbcde和foki-cas8进行重构。
[0680]
为了进行dna切割测定,反应混合物如下所示。将包含具有30bp间隔区间的j3/l3双靶标序列(表30中的seq id no:483)的质粒底物与不同浓度的foki-cascade复合体(3-100nm)在质粒dna最终浓度为13.3ng/μl的1x cascade切割缓冲液中的15μl反应物中一起孵育。将反应物在37℃下孵育30分钟,然后加入3μl 6x sds载样染料。添加载样染料以使结合的foki-cascade复合体变性。通过0.8%琼脂糖凝胶电泳分离反应混合物组分。电泳后,用sybrtm安全dna凝胶染色剂(thermo scientific,wilmington,de)对凝胶染色。
[0681]
作为阳性对照,用靶向cascade j3靶序列的20bp部分(sgrna-j3;间隔区序列示出为seq id no:501)的单向导rna(sgrna)将酿脓链球菌cas9蛋白程序化。通过在1x cce缓冲液(20mm hepes ph 7.4,10mm mgcl2,150mm kcl,5%甘油)中混合cas9与2倍摩尔过量的sgrna来重构cas9/sgrna-j3复合体。通过在37℃下孵育反应物30分钟,在相同浓度范围(3
–
100nm)内评估了此cas9/sgrna-j3复合体的切割。实验中还包括含有未切割质粒dna的对照泳道,以及用nhei限制酶(new england biolabs,ipswich,ma)线性化的质粒dna。靶标dna切割可以通过质粒中的迁移率变化来证明,因为未切割的质粒dna是超螺旋的,并且具有比切割的线性化质粒dna更快的迁移率。带切口的开环质粒dna具有比超螺旋和线性化质粒dna都慢的迁移率。
[0682]
从这些实验获得的数据表明,在浓度范围内,foki-cascade复合体表现出与cas9-sgrna类似的靶标dna切割活性。在最高测试浓度(100nm)下,通过foki-cascade复合体和cas9-sgrna对质粒靶标进行定量线性化。
[0683]
还测试了foki-cascade复合体试剂的靶标dna切割的动力学。将含有具有30bp间隔区间的j3/l3双靶序列(seq id no:483)的质粒底物与200nm foki
–
cascade复合体或200nm cas9
–
sgrna一起在质粒dna终浓度为13.3ng/μl的15μl反应物中孵育。在0、7、10、15、20、25或30分钟时将反应淬灭,并且按如上所述通过琼脂糖凝胶电泳分离反应物组分。foki
–
cascade复合体表现出与cas9/sgrna-j3复合体相似但稍微更慢速率的靶标dna切割活性,其中对于foki-cascade复合体,到25分钟时间点时靶标质粒被定量线性化,且对于cas9/sgrna-j3复合体,为20分钟时间点。
[0684]
还测试了foki-cascade复合体试剂对pacyc-duet1非靶标质粒底物的非特异性dna切割和/或切口活性,相对于j3/l3双靶标质粒底物的特异性dna切割。表32包含用于该对照的pacyc-duet1非靶标质粒底物的序列(seq id no:502)。具体地,研究了非特异性和特异性dna靶标切割的依赖性作为反应缓冲液中一价盐浓度的函数。制备了1x cascade切割缓冲液(20mm tris-cl,ph 7.5,200mm nacl,5mm mgcl2,1mm tcep和5%甘油)的变型,其中nacl浓度从200mm降至150mm、100mm或50mm,并通过将200nm foki-cascade复合体与13.3ng/μl的j3/l3靶标质粒或13.3ng/μl的pacyc-duet1非靶标质粒一起孵育进行了如上所述的相同的切割反应。进行了另外的对照反应,其中nacl的浓度保持在100mm,但是用
10mm edta取代5mm mgcl2,由于foki需要二价金属离子用于dna切割,因此其预期会取消切割。因此,对非靶标质粒和j3/l3靶标质粒进行以下反应条件:
–
foki-cascade复合体;+foki-cascade复合体,100mm nacl缓冲液+10mm edta;+foki-cascade复合体,50mm nacl缓冲液;+foki-cascade复合体,100mm nacl缓冲液;+foki-cascade复合体,150mm nacl缓冲液;+foki-cascade复合体,200mm nacl缓冲液。数据证明foki-cascade复合体显示在低盐浓度<200mm nacl下非特异性切口于非靶标和j3/l3靶标质粒,但在200mm nacl的一价盐浓度下,非靶标质粒保持完整,但j3/l3靶标质粒被定量线性化。此外,缓冲液包含edta引起靶标切割的完全取消,如同预期的一样。
[0685]
为了确认foki
–
cascade复合体在预期位置,即在分隔j3和l3靶标的间隔区间序列的中间内切割靶标质粒,进行了一项实验,其中首先将靶标质粒与foki-cascade复合体一起孵育,然后与afei限制酶(new england biolabs,ipswich,ma)一起孵育,该酶在质粒底物中的其他地方切割。因此,通过foki-cascade 1复合体和afei进行的切割将超螺旋的环形质粒转换成两个线性片段,在琼脂糖凝胶上作为不同种类迁移。具体而言,预期切割会产生长度为2427bp和1357bp的片段。
[0686]
将13.3ng/μl j3/l3靶标质粒与200nm foki-cascade 1复合体一起孵育30分钟,然后将1μl afei(10单位/μl;new england biolabs,ipswich,ma)加入反应物中,然后在37℃下再另外孵育30分钟。如上所述,将反应产物通过琼脂糖凝胶电泳分离。另外,为了进行对照实验,将靶标质粒仅与foki-cascade 1复合体或仅afei一起孵育,并且用可被afei切割但不能由foki-cascade 1复合体切割(因为质粒缺少j3/l3双靶标)的非靶标质粒进行相同的反应。表32包含用于该对照的pacyc-duet1非靶标质粒底物的序列(seq id no:502)。因此,将非靶标质粒和j3/l3靶标质粒进行以下反应条件:
–
afei/
–
foki-cascade复合体;
–
afei/+foki-cascade复合体;+afei/+foki-cascade复合体;和+afei/
–
foki-cascade复合体。数据表明foki-cascade复合体在预期的位置切割了靶标质粒,因为与foki-cascade 1复合体和afei一起孵育会产生两种预期长度的线性产物。
[0687]
为了进一步确定由foki-cascade复合体进行的dna切割的序列特异性,生成了包含以下突变的另外的对照质粒底物:位于j3靶标两侧的pam中的突变、位于l3靶标两侧的pam中的突变、位于j3/l3靶标两侧的两个pams中的突变;j3靶标内的间隔区序列中的突变、l3靶标中的间隔区序列中的突变、j3/l3靶标;和j3靶标但不是l3靶标、l3靶标但不是j3靶标,以及既非j3也非l3靶标内的两个间隔区序列中的突变。因此,质粒底物如下:j3 pam突变体、l3 pam突变体、j3/l3 pam突变体、j3间隔区突变体、l3间隔区突变体、j3/l3间隔区突变体、非靶标质粒、仅j3靶标、仅l3靶标和j3/l3靶标质粒。对每种靶标进行以下反应条件:
–
ndei/
–
foki-cascade复合体;+ndei/
–
foki-cascade复合体;和
–
ndei/+foki-cascade 1复合体。表32包含上述所有突变的质粒底物(seq id no:502至seq id no:510)的序列。
no:500中描述。表33包含靶向hsa01至hsa16 gdna序列的每对中两种crrnas的序列;crrna的间隔区带有下划线且小写,并且向导区域的5'和3'序列与来自crispr阵列的重复序列相对应。
[0693]
[0694]
[0695][0696]
纯化16种foki
–
cascade复合体后,按上述方法进行切割反应,其中将foki-cascade复合体与含有智人基因组位点hsa01至hsa16的质粒底物一起孵育,并通过琼脂糖凝胶电泳分离出反应产物。数据表明,在16种rnp试剂中,14/16种(hsa03
–
hsa16)表现出几乎定量的dna切割,如通过将超螺旋的圆形质粒底物转化为切割的线性形式所证明的。仅构建体hsa01和hsa02显示出部分切口活性。此外,数据表明,使用设计的16种成对的grnas可以有效地将foki-cascade复合体程序化以靶向与治疗相关的智人基因。
[0697]
实施例8
[0698]
将foki-cascade rnp复合体引入靶标细胞中
[0699]
本实施例示出了包含foki融合蛋白以促进在人类细胞中的基因组编辑的大肠杆
菌i-e型cascade复合体的设计和和递送,并描述了其作为预组装的cascade rnp复合体递送至靶标细胞中。
[0700]
a.包含foki以用于转化至细胞中的cascade rnp复合体的产生
[0701]
最小crispr阵列被设计为靶向人类基因组中的8种不同的位点。每个最小crispr阵列包含两个间隔区序列,其两侧均为crispr重复序列。两个间隔区序列靶向基因中间隔30bp的位点(即,30bp间隔区间区域),并且每个间隔区被设计为结合临近靶标细胞基因组中的aag或atg前间区序列邻近基序(pam)序列的靶序列。通过将退火的寡核苷酸(integrated dna technologies,coralville,ia)连接至用于细菌表达的pacyc-duet1(milliporesigma,hayward,ca)在载体骨架中而产生了包含每种最小crispr阵列的质粒载体。
[0702]
用于产生最小crispr阵列中选择的间隔区的重叠的引物示出在表34中,并且引物序列描述于表35中。
[0703][0704]
[0705]
[0706][0707]
实施例2中详细描述了用于产生cascade rnp复合体的细菌表达载体的设计。简而言之,每个cas基因都由单个操纵子表达,并且cas基因的编码序列按cas8
–
cse2
–
cas7
–
cas5
–
cas6的顺序排列。foki部分通过30-aa连接子附接到cas8,并且核定位信号(nls)被附
接到foki-cas8(foki-cascade复合体)的n端和cas6(以下称为foki-cascade-nls-cas6复合体,seq id no:577)的n端。
[0708]
基本上按实施例5a中所述从大肠杆菌将foki-cascade-nls-cas6复合体纯化为组装的复合体。
[0709]
b.将包含foki的cascade rnp复合体转染至真核细胞中
[0710]
在37℃、5%co2和100%湿度下,将hek293细胞(atcc,manassas,va)培养在补充有10%fbs和1x抗生素-抗真菌溶液(mediatech,inc.,manassas,va)的dmem培养基中的悬浮液中。使用96孔shuttle系统(lonza,allendale,nj)转染hek293细胞。在进行核转染之前,将5μl foki-cascade rnps转移到96孔板的单个孔中。每个孔中包含~225-500pmol foki-cascade-nls-cas6复合体,具体取决于rnp。将hek293细胞转移至50ml锥形离心管中,并以200xg离心3分钟。吸出培养基,并在不含钙和镁的pbs中洗涤细胞沉淀。将细胞再次离心一次并以1x107个细胞/ml的浓度重悬于nucleofector sf(lonza,allendale,nj)缓冲液中。将20μl该细胞悬液添加到于96孔板中的foki-cascade-nls-cas6复合体中,混合,然后将全部体积转移到96孔nucleocuvette
tm
(lonza,allendale,nj)板中。然后将板装载到nucleofector
tm
96孔shuttle
tm
系统(lonza,allendale,nj)中,并使用96-cm-130nucleofector
tm
程序(lonza,allendale,nj)对细胞进行核转染。核转染后,立即将80μl完全dmem培养基添加到96孔nucleocuvette
tm
(lonza,allendale,nj)板的每个孔中。然后将孔的全部内容物转移至包含100μl完全dmem培养基的96孔组织培养板中。将细胞在37℃、5%co2和100%湿度下培养~72小时。
[0711]
~72小时后,将hek293细胞以500xg离心5分钟,然后除去培养基。在无钙和无镁的pbs中洗涤细胞。然后将细胞沉淀重悬于50μl quickextract dna提取溶液(epicentre,madison,wi)中。然后将获得的gdna样品在37℃下孵育10分钟,在65℃下孵育6分钟,并在95℃下孵育3分钟以终止反应。然后将gdna样品用50μl水稀释,并保存在-20℃下用于后续的深度测序分析。
[0712]
c.来自转染细胞的gdna的深度测序
[0713]
使用分离的gdna,利用1x浓度的q5热启动高保真2x预混液(new england biolabs,ipswich,ma)、于10μl最终体积中的0.5μm的每种引物、3.75μl的gdna进行第一次pcr,并在98℃下扩增1分钟,35个循环的98℃下扩增10秒、60℃下扩增20秒、72℃下扩增30秒,以及在72℃下最终延伸2分钟。将pcr反应物在水中以1:100稀释。表36显示了靶标特异性的引物。靶标特异性引物包含与illumina相容的序列,使得可以使用miseq测序仪(illumina,san diego,ca)分析扩增的产物。
rnp预期切割位点周围10bp)的读段归类,并记录。
[0725]
将总插入缺失读段除以野生型读段和插入缺失读段之和,以给出突变读段的百分比。
[0726]
图28显示了基因组编辑(图28,垂直轴,“%编辑”)作为foki-cascade-nls-cas6复合体核转染(n=1)的函数(图27,水平轴,hsa3、hsa4、hsa5、hsa6、hsa7、hsa8、hsa9和hsa10)。在图28中,空心条是阴性对照,并且黑色条是添加的foki-cascade-nls-cas6复合体)。foki-cascade-nls-cas6复合体在所有8个位点处诱导了编辑。编辑范围为~0.2-5%插入缺失,并且插入缺失集中在预测的切割位点周围,在间隔区间区域的中间。
[0727]
实施例9
[0728]
将foki-cascade rnp复合体的质粒编码组分引入靶标细胞中
[0729]
本实施例示出了包含foki融合蛋白以促进在人类细胞中的基因组编辑的大肠杆菌i-e型cascade复合体的设计和递送。本实施例还描述将表达cascade复合体组分的质粒载体递送至真核细胞中。
[0730]
a.待转染至靶标细胞中的编码foki-cascade rnp组分的载体的产生
[0731]
设计了最小crispr阵列以靶向人类基因组中的trac位点。最小crispr阵列包含两个间隔区序列,其两侧均是crispr重复序列,如实施例1和3中所述的。两个间隔区序列靶向基因组中间隔30bp的位点,并且每个间隔区与临近aag pam序列的基因组序列互补。通过将退火的编码两个间隔区序列两侧的crispr重复的寡核苷酸(integrated dna technologies,coralville,ia)连接至具有两个crispr重复序列的哺乳动物表达载体中,而产生了包含最小crispr阵列的质粒载体。所得的质粒包含表达来自人u6(hu6)启动子(seq id no:454)的两种向导的“重复区-间隔区-重复区-间隔区-重复区”。
[0732]
将foki-cascade rnp蛋白组分编码基因克隆至包含cmv启动子的质粒载体中,以使得能够在哺乳动物细胞中递送和表达。将cas基因克隆至单独的质粒(seq id no:448至seq id no:451和seq id no:453)中或单个的质粒中作为多顺反子构建体,其中每个基因经由2a病毒肽“核糖体跳跃”序列连接(在seq id no:455中)。经由两种不同的方法将foki-cascade rnp复合体递送至真核细胞中:cas基因和最小crispr阵列提供在单独的质粒(六质粒递送系统,seq id no:448至seq id no:451、seq id no:453和seq id no:454)上,或编码所有cas基因的一个质粒上作为多顺反子构建体,以及编码最小crispr阵列的第二质粒(双质粒递送系统,seq id no:454和seq id no:455)上。
[0733]
b.编码foki-cascade rnp复合体的质粒的转染
[0734]
按实施例8b中详细描述的对六质粒递送系统和双质粒递送系统进行转染条件,进行了以下修改。进行核转染前,将5μl质粒载体溶液转移至96孔板的单个孔中。最初通过检查用于基因组编辑的每个组件的必要性对六质粒递送系统进行了测试。更具体地,将质粒“混合物(cocktails)”添加到每个孔中,使得存在恒定数量(420ng)的五种质粒和可变数量的第六种质粒(0ng、70ng、700ng或1400ng)。接下来,通过以固定量(3.5μg)的总质粒dna进行核转染,同时改变最小crispr阵列质粒与cas编码质粒的比例,比较了六质粒递送系统和双质粒递送系统。最后,在核转染后~72小时收获裂解物,以用于随后的深度测序分析。
[0735]
c.来自转染细胞的gdna的深度测序和数据分析
[0736]
按实施例8c中详细描述的进行深度测序,但仅使用来自表36的靶标特异性引物y
和z。
[0737]
d.深度测序数据分析
[0738]
按实施例8d中详细描述的进行深度测序数据分析。图29显示了trac位点处的基因组编辑(图29,垂直轴,“%编辑”)作为六质粒递送策略(n=1)中每种foki-cascade组分的函数(图29,水平轴,向导、foki-cas8、cse2、cas7、cas5、cas6和参照样本)。在图29中,空心条代表0ng的foki-cascade组分,斑点条代表70ng的foki-cascade组分,方形图案条代表700ng的foki-cascade组分,并且条纹条代表1,400ng的foki-cascade组分(对于每种foki-cascade组分,在水平轴上的条顺序分别为从左至右)。如图所示的,如果缺少给定的组件,将取消或大幅减少编辑(在cse2的情况下)。这确认了每种cascade组件对于经由质粒递送进行编辑都是必需的。
[0739]
图30显示了将利用六质粒递送系统或双质粒递送系统的基因组编辑进行比较的数据。图30显示了靶标基因位点处的基因组编辑(图30,垂直轴,“%编辑”)作为不同浓度的六质粒(图30,空心条)和双质粒(图30,黑条)系统的每种组分的函数(图30,水平轴上的条顺序从左至右分别是六质粒系统和双质粒系统)。沿水平轴的数字分组指的是组分的量:顶线=ng的总质粒,第二条线=ng的最小crispr阵列质粒,并且第三条线=ng的cas编码质粒(例如,第一数字分组:顶线=总质粒,3500ng;第二条线=最小crispr阵列质粒,0ng;并且第三条线=cas编码质粒,3500ng)。
[0740]
在这两种方法中,利用最高比例的cas:最小crispr阵列质粒实现了最高水平的编辑。另外,多顺反子质粒使得能够进行更高水平的编辑,这可能是由于每μg质粒的转录增加所致。
[0741]
实施例10
[0742]
循环排列的cascade亚基蛋白
[0743]
本实施例示出了使用结构引导的建模方法的循环排列的(cp)大肠杆菌i-e型cas7蛋白的计算机设计、克隆、表达和纯化。
[0744]
a.计算机设计
[0745]
基于大肠杆菌cascade晶体结构5h9e.pdb(www.rcsb.org/pdb/;hayes,r.p,et al.,nature 530(7591):499-503(2016)),使用结构引导的方法将大肠杆菌i-e型cas7蛋白(seq id no:18)环状排列。利用具有序列甘氨酸-丝氨酸(g-s)的双氨基酸肽连接子连接天然的cas7 n端和c端。在与野生型cas7多肽序列中的残基301和302之间的肽键相对应的位置处开放该循环的cas7的多肽序列,以形成新的n端(残基302)和新的c端(残基301),导致循环排列形式的cas7蛋白(cp-cas7 v1蛋白)。新的n端和新的c端的设计位置使其与融合蛋白或连接子区域连接,而不会干扰cas7蛋白折叠或cascade复合体组装。将甲硫氨酸残基添加到cp-cas7 v1蛋白(seq id no:578)的新的n端(即,与野生型cas7蛋白的残基302相对应的氨基酸残基)。
[0746]
使用g-s连接子对第二种cp-cas7蛋白——cp-cas7 v2蛋白进行了类似的改造。cp-cas7 v2蛋白的n端和c端分别对应于野生型cas7序列中的残基338和339。新的n端和新的c端的设计位置使其与融合蛋白或连接子区域连接,而不会干扰cas7蛋白折叠或cascade复合体组装。将甲硫氨酸残基添加到cp-cas7 v2蛋白(seq id no:579)的n端(即,对应于野生型cas7蛋白的残基339的氨基酸残基)。
[0747]
b.包含cp-cas7的cascade复合体的克隆、表达和纯化
[0748]
对cp-cas7 v1蛋白和cp-cas7 v2蛋白的计算机设计的多肽序列的dna编码序列进行了密码子优化,以用于在大肠杆菌中表达。
[0749]
提供这些dna编码序列给商业制造商(genscript,piscataway,nj)进行合成。将dna序列单独引入cascade-操纵子表达载体(表19;seq id no:441)中,以取代表达载体中的野生型cas7蛋白,如实施例2中所述的。
[0750]
利用编码表20中所示的j3靶标(seq id no:444)的向导rna的第二载体,将每种表达载体转染至大肠杆菌bl21 startm(thermo fisher scientific,waltham,ma)细胞中,如实施例2中所述的。按实施例4b中所述的培养细胞。按实施例5a中所述的纯化包含cas5、cas6、cp-cas7 v1、cse2和cas8蛋白,以及向导rna/靶标j3;和cas5、cas6、cp-cas7 v2、cse2和cas8蛋白以及向导rna/靶标j3的大肠杆菌i-e型cascade复合体。
[0751]
包含cp-cas7变体的cascade复合体的纯化表明,循环排列的i-e型crispr-cas亚基蛋白质可以成功用于形成形成与包含野生型蛋白的cascade复合体具有基本上相同的组合物(基于分子量)的cascade复合体。
[0752]
c.cascade/cp-cas7和j3靶标的emsa(电泳迁移率变动测定)
[0753]
按本实施例所述纯化纯化的cascade/cp-cas7复合体,并对其进行emsa,以证明与其各自靶序列的特异性结合。简而言之,将cascade/cp-cas7和cascade/wt-cas7纯化并浓缩至10mg/ml。cy5双链靶标dna基本按照实施例6a所述制备,并在te缓冲液中稀释至1μm(j3靶标seq id no:469和seq id no:472以及ccr5靶标seq id no:474和seq id no:470)。将cascade复合体和标记的双链靶标dna以不同的蛋白质/靶标比例于37℃下孵育30min。孵育后,立即将2μl 50%甘油添加到样品中,并将其加载到5%天然paa凝胶上。凝胶在0.5xtbe缓冲液中于4℃在70v下运行90min,并在azure c600 bioimager(azure biosystems,dublin,ca)上成像,并对条带进行定量。数据在表37中提供。
[0754][0755]
*lod=低于检测限
[0756]
实施例11
[0757]
cascade亚基融合蛋白
[0758]
a.cascade亚基与foki融合
[0759]
本实施例示出了融合至foki核酸酶域以向cascade复合体赋予核酸酶活性的大肠杆菌i-e型cas8蛋白的计算机设计、克隆、表达和纯化。
[0760]
大肠杆菌i-e型cas8在末端与海床黄杆菌(flavobacterium okeanokoites)foki核酸酶域(genbank no.aaa24927.1)融合。foki核酸酶域包含由guo,et al.(guo,j.,et al.,j.mol.biol.400:96-107(2010))描述的sharkey变体中包含的残基,且在均二聚化作用后催化双链dna切割。foki核酸酶的氨基酸序列(seq id no:580)包含残基q384至f579(genbank编号aaa24927.1),并具有以下电突变:e486q、l499i和d469n。简言之,使用连接子序列(seq id no:582)将foki sharkey核酸酶域(seq id no:581)n端融合至cas8。为了纯化目的,先用六组氨酸标签(his6,seq id no:583),再用mbp标签(seq id no:584),再用tev蛋白酶切割序列(seq id no:585)、核定位信号(nls,seq id no:586)和ggs连接子n端附接到foki的残基384上。最终的构建体在蛋白质序列中包含nh3-his6-mbp-tev-nls-ggs-fokisharkey-30aa-连接子-cas8-cooh(seq id no:413)。
[0761]
将计算机设计的dna序列提供给商业制造商(genscript,piscataway,nj)进行合成。将dna序列克隆到pet表达(milliporesigma,hayward,ca)家族载体骨架中,由于如实施例2中所述的kanr基因的存在,该载体骨架赋予了卡那霉素抗性,导致携带nh3-his6-mbp-tev-nls-ggs-fokisharkey-30aa-连接子-cas8-cooh(seq id no:439)的载体。
[0762]
按实施例4b和实施例5c中所述的对大肠杆菌i-e型cascade h3-his6-mbp-tev-nls-ggs-fokisharkey-30aa-连接子-cas8-cooh(seq id no:439)进行表达和纯化。tev切割后的蛋白序列包含nh3-nls-ggs-fokisharkey-30aa-连接子-cas8-cooh(seq id no:587)。
[0763]
类似地,按实施例1和2(seq id no:442)所述的,在携带nls-foki-连接子-cas8_his6
–
hrv3c
–
cse2_cas7_cas5_cas6的载体中构建了fok1-cas8融合蛋白。利用编码j3靶标(seq id no:444)的向导rna的第二载体,将每种表达载体转染至大肠杆菌bl21 startm(thermo fisher scientific,waltham,ma)细胞中,如实施例2中所述的。按实施例4b和实施例5a中所述表达和纯化该构建体。包含融合的foki-cas8变体的cascade复合体的纯化表明,核酸酶融合的i-e型crispr-cas亚基蛋白质可成功用于形成与包含野生型蛋白的cascade复合体具有基本相同的组合物(基于分子量)的cascade复合体。foki-cas8融合物已成功用于靶标核酸的生化切割(实施例7)和真核细胞中基因组序列的细胞内切割(实施例8d和实施例9d)。
[0764]
表38列出了cas亚基蛋白-酶融合的其他实例。在表38中,apobec对应于胞苷脱氨酶途径成员的基因(人类apobec i genbank编号ab009426、人类apobec 3f genbank编号ch471095、人类apobec 3g genbank编号cr456472、大鼠apobec ucsc基因组阅读器id rgd:2133大鼠);aid对应于活化诱导的胞苷脱氨酶(genbank编号ay536516);aid直系同源中的pmcda1(参见,例如nishida,et al.,science 16:353(2016);iwamatsu,et al.,j.biochem.110:151-158(1991));pvuiihifit46g是pvuii高保真度变体t46g(参见,例如fonfara,et al.,nucleic acids res.40:847-860(2012));pvuii单链t46g描述于pdbid 3ksk中);i-tevi是来自噬菌体t4的位点特异性的、序列耐受性的归巢核酸内切酶,并且包含n端催化域以及c端dna结合域(该域通过长的、柔性的连接子连接)(参见,例如van roey,et al.,embo j.20:3631-3637(2001));bcni(参见,例如sokolowska,et al.,j.mol.biol.369:722-734(2007));和mvai(参见,例如kaus-drobek,et al.,nucleic acids res.35:2035-2046(2007))是限制酶。
[0765][0766][0767]
b.与另一cascade亚基蛋白的cascade亚基蛋白融合
[0768]
基于大肠杆菌cascade晶体结构5h9e.pdb(www.rcsb.org/pdb/;参见,例如hayes,r.p,et al.,nature 530(7591):499-503(2016)),使用结构引导的方法将cascade复合体的两种cse2蛋白融合在一起。简言之,使用10-aa柔性连接子(seq id no:589),将一个cse2的c端和第二cse2的n端融合在一起。cse2-cse2(casb_casb)融合蛋白的完整序列示出在seq id no:588中。
[0769]
将计算机设计的dna序列提供给商业制造商(genscript,piscataway,nj)进行合成。将dna序列克隆到实施例2中设计的表达载体(seq id no:441)中。将cse2序列用seq id no:588交换。
[0770]
利用编码j3靶标(seq id no:444)的向导rna的第二载体将每种表达载体转染至大肠杆菌bl21 startm(thermo fisher scientific,waltham,ma)细胞中,如实施例2中所
述的。按实施例4b和5b中所述表达和纯化包含cas5、cas6、cas7、cse2-cse2和cas8的大肠杆菌i-e型cascade复合体。包含融合的cse2-cse2变体的cascade复合体的纯化证明融合的i-e型crispr-cas亚基蛋白成功地形成了cascade复合体与cascade复合体包含野生型蛋白具有基本上相同的组合物(基于分子量)。
[0771]
c.cascade/cse2-cse2和j3靶标的电子迁移率变动测定(emsa)
[0772]
按本实施例中所述纯化纯化的cascade/cse2-cse2复合体,并对其进行emsa,以证明与其各自靶序列的特异性结合。简而言之,将cascade/se2-cse2和cascade/wt-cse2纯化并浓缩至10mg/ml。按实施例6a中所述制备cy5双链靶标dna,并在te缓冲液中稀释至1m(j3靶向seq id no:469,并且seq id no:472和ccr5靶向seq id no:474和seq id no:470)。将cascade复合体和标记的双链靶标dna在37℃下以不同的蛋白质/靶标比例孵育30min。孵育后,立即将2μl 50%甘油添加到样品中,并将其加载到5%天然paa凝胶上。将凝胶在0.5xtbe缓冲液中于4℃在70v下运行90min,并在azure c600 bioimager(azure biosystems,dublin,ca)上成像,并对条带进行定量。数据提供在表39中。
[0773][0774]
*lod=低于检测限
[0775]
d.与另一cascade亚基蛋白和酶促蛋白域融合的cascade亚基蛋白
[0776]
选择胞苷脱氨酶rapobec1(载脂蛋白b mrna编辑酶催化亚基1,褐家鼠(rattus norvegicus);ncbi基因id:25383,uensembl:ensrnog00000015411)用于融合。使用结构引导的方法,基于大肠杆菌cascade晶体结构5h9e.pdb(www.rcsb.org/pdb/;参见,例如hayes,r.p,et al.,nature 530(7591):499-503(2016))将cse2-cse2蛋白与rapobec1融合。简而言之,使用9-aa柔性连接子(seq id no:591)将rapobec1(seq id no:590)的c端融合至cse2-cse2二聚物(上述的)的n端。rapobeci_cse2-cse2融合蛋白的完整序列示出在seq id no:592中。
[0777]
将计算机设计的dna序列提供给商业制造商(genscript,piscataway,nj)进行合成。将dna序列克隆至表达载体(seq id no:441)中,取代cse2序列。利用编码j3靶标(seq id no:444)的向导rna的第二载体将每种表达载体转染至大肠杆菌bl21 startm(thermo fisher scientific,waltham,ma)细胞中,如实施例2中所述的。按实施例4b和5b中所述表达和纯化包含cas5、cas6、cas7、rapobec1_cse2-cse2和cas8的大肠杆菌i-e型cascade复合体。包含融合的rapobec1_cse2-cse2变体的cascade复合体的纯化证明,与i-e型crispr-cas亚基蛋白的胞苷脱氨酶融合被成功地用于形成cascade复合体与包含野生型蛋白的cascade复合体具有基本上相同的组合物(基于分子量)。表40提供了与cse2-cse2的酶融合的实例。
[0778][0779]
实施例12
[0780]
与转录激活/抑制域的cascade亚基蛋白融合
[0781]
本实施例示出了融合至vp64活化域以向cascade复合体赋予转录激活活性的大肠杆菌i-e型cp-cas7蛋白的设计。
[0782]
vp64是一种包含利用甘氨酸-丝氨酸(gs)连接子连接的4个串联拷贝的vp16(单纯疱疹病毒蛋白16,dalddfdldml(seq id no:614);氨基酸437-447,uniprot:ul48)的转录活化剂。当融合至可以结合基因启动子附近的蛋白域时,vp64(seq id no:615)用作强转录活化剂。大肠杆菌i-e型cp-cas7 v2(seq id no:616)可以选择用于改造。
[0783]
活化域vp64可以融合至cpcas7 v2的n端(实施例10a所述的)。可以选择连接子(例如,5-50个氨基酸的长度)以操作性地连接cpcas7 v2和vp64域。
[0784]
可以将计算机设计的dna序列提供给商业制造商进行合成。可以将编码vp64-cpcas7 v2融合蛋白的dna序列克隆至表达载体(例如,seq id no:455,其中vp64-cpcas7 v2可用于取代cas7)中。可以利用编码j3靶标(seq id no:444)的向导rna的第二载体将每种表达载体转染至大肠杆菌bl21 startm(thermo fisher scientific,waltham,ma)细胞中,如实施例2中所述的。可以按实施例4和5中所述表达和纯化包含cas5、cas6、vp64_cpcas7 v2、cse2和cas8的大肠杆菌i-e型cascade复合体。包含融合的vp64_cpcas7 v2变体的cascade复合体的纯化可以用于形成与包含野生型蛋白的cascade复合体具有基本上相同的组合物(基于分子量)的cascade复合体。
[0785]
靶向特定基因的启动子区域的向导的选择可以用于验证包含融合的vp64_cpcas7 v2的cascade复合体促进基因转录激活的能力。
[0786]
实施例13
[0787]
融合至cascade亚基的功能域被dcas9/向导复合体的位点定向募集
[0788]
本实施例描述了利用用于将融合至功能域的一种或多种cascade亚基蛋白(即cas6、cas5等)募集至ii型crisprcas蛋白/向导rna复合体结合位点的第一类i型crispr重复茎序列(例如,i-f型crispr重复茎序列)改造2类ii型crisprsgrna、crrna、tracrrna或crrna和tracrrna序列的方法。这里的该方法自gilbert,l.,et.al.,cell 154(2):442-451(2013)和ferry,q,et.al.,nature communication 8:14633doi:10.1038/ncomms14633
(2017)调整而来。
[0789]
a.改造ii型向导rna
[0790]
可以选择ii型crisprsgrna、crrna、tracrrna或crrna和tracrrna(统称为“ii型向导rna”)用于改造。
[0791]
可以利用计算机评估ii型向导rna序列的掺入的i型crispr重复茎序列的区域。can be attached在ii型向导rna的5
’
或3
’
端处,ii型向导rna的内部附接i型crispr重复茎序列,或者可以取代ii型向导rna中的二级结构(例如,3
’
发卡元件)。i型crispr重复茎序列的掺入可以伴随有连接子元件核苷酸序列。3
’
改造以包含i型crispr重复茎序列的ii型tracrrna的实例提供在表41中。
[0792][0793]
*i型crispr重复茎序列带下划线且为小写字母。相应的dna编码序列提供为seq id no:618。
[0794]
可以选择哺乳动物的基因,如c-x-c趋化因子受体4型(cxcr4)进行靶向。可以在计算机上扫描5'utr和外显子1之间的连接处ii型crisprcas蛋白pam序列附近存在的ii型crisprcas蛋白靶序列(例如,5
’-
ngg)。可以将5
’
方向上游存在的20个核苷酸靶序列掺入到ii型crrna中。表42中显示了靶向cxcr4的ii型crrna的实例。
[0795][0796]
*相应的dna编码序列提供为seq id no:620。
[0797]
可选地,靶向间隔区(rna)(seq id no:619)的cxcr4的3'端可以利用连接子与3
’
i型crispr重复茎序列(rna)(seq id no:617)一起共价连接至ii型tracrrna的5'端。合适的连接子元件是5
’-
gaaa-3
’
。
[0798]
可以将具有掺入的i型crispr重复茎序列的ii型向导rnas提供给商业制造商进行合成。
[0799]
可以将i型cascade亚基蛋白(例如,cas6)操作性地连接至转录激活或抑制域(例如,krab),并利用如实施例12中所述的核定位信号(nls)进行c端标记。
[0800]
可以将ii型cas蛋白(例如,cas9)突变,使得其被催化失活(例如dcas9)并用nls序列标记。
[0801]
可以从大肠杆菌重组表达和纯化cas6-krab-nls蛋白和dcas9-nls蛋白。
[0802]
可以以60pmol dcas9蛋白:60pmol cas6-krab-nls:120pmol:靶向crrna的cxcr4:
120pmol tracrrna 3
’
改造以包含i型crispr重复茎序列的浓度形成rnp复合体。在利用dcas9和cas6-krab-nls组装之前,可以在2μl的最终体积中将120pmol靶向crrna的cxcr4和120pmol tracrrna 3
’
改造以包含i型crispr重复茎序列(本文中称为“改造的ii型向导rna”)中的每种稀释至所需的总浓度(120pmol),在95℃下孵育2分钟,从热循环仪移除,并允许平衡至室温。可以将dcas9和cas6-krab-nls蛋白在结合缓冲液(20mm hepes,100mm kcl,5mm mgcl2和5%甘油,ph 7.4)中稀释至合适的浓度,至最终体积为3μl,并与2μl ii型rna混合,然后在37℃下孵育30分钟。可以将未转染的对照(例如,仅缓冲液)、未改造的ii型向导rna或未连接至抑制域的cas6用于组装阴性对照rnps。
[0803]
b.使用dcas9:cas6-krab-nls:改造的ii型向导rna进行细胞转染
[0804]
可以使用96孔shuttle系统(lonza,allendale,nj)和以下方案将dcas9:cas6-krab-nls:改造的ii型向导rna核蛋白复合体转染到hek293细胞(atcc,manassas va):复合体可以以5μl的最终体积分配到96孔板的单个孔中。可以从hek293细胞培养板上除去细胞培养基,并用tryple
tm
(thermo scientific,wilmington,de)分离细胞。可通过以200x g离心3分钟沉淀出悬浮的hek293细胞,吸出tryple试剂,并用无钙和无镁磷酸盐缓冲盐水(pbs)洗涤细胞。可以通过以200x g离心3分钟来沉淀细胞,吸出pbs,然后将细胞沉淀重悬于10ml不含钙和镁的pbs中。
[0805]
可以使用ii自动化细胞计数器(life technologies;grand island,ny)对细胞进行计数。可将2.2x 107个细胞转移到1.5ml微量离心管中并沉淀。可以吸出pbs,并将细胞重悬在nucleofector
tm
sf(lonza,allendale,nj)溶液中,至密度为1x 107个细胞/m。然后可以将20μl的细胞悬浮液添加到每个含有5μl rnp复合体的单个孔中,然后将来自每个孔的全部体积转移到96孔nucleocuvette
tm
(lonza,allendale,nj)板的孔中。可以使用96-cm-130nucleofector
tm
(lonza,allendale,nj)程序将板装载到nucleofector
tm
96孔shuttle
tm
(lonza,allendale,nj)上并对细胞进行核转染。核转染后,可以向每个孔添加补充10%胎牛血清(fbs;thermo scientific,wilmington,de)、青霉素和链霉素(life technologies,grand island,ny)的70μl dulbecco改良的eagle培养基(dmem;thermo scientific,wilmington,de),并可以将50μl的细胞悬浮物转移至含有150μl预热的dmem完全培养基的96孔细胞培养板中。可以将板转移到组织培养孵育器中,并在37℃下于5%co2中保持48小时。
[0806]
在dcas9:cas6-krab-nls:改造的ii型向导rna核蛋白复合体进行核转染后72小时,可以评估细胞对cxcr4表达的抑制作用。可以从hek293吸出培养基,并用无钙和无镁的pbs洗涤细胞一次,然后通过加入tryple(life technologies,grand island,ny)进行胰蛋白酶消化,然后在37oc下孵育3-5分钟。可以将胰蛋白酶消化的细胞轻轻上下吸取以形成单细胞悬液,然后可以通过以200x g离心3分钟来沉淀细胞。离心后,可将培养基吸出,并将细胞重悬于10mm edta/pbs缓冲液中,并轻轻混合成单细胞悬液。可以在室温下,在含有10%fbs的pbs中,使用与抗人cxcr4抗体(medical&biological laboratories co.,nagoya,japan)缀合的0.05%fitc将单细胞悬液染色1小时。同种型对照和天然rnp对照也可以进行类似染色以供参考。然后可以对染色的细胞进行分选lsr ii流式细胞仪(bd laboratories,san jose,ca),并记录fitc阳性荧光细胞群。
[0807]
通过检测的dcas9:cas6-krab-nls:改造的ii型向导rna核转染样品的荧光相比非
转染对照的荧光检测值的降低来测量cxcr4表达的降低。来自流式细胞仪的荧光的降低可用于证明具有i型crispr重复茎序列的改造的ii型向导rna可与核酸酶缺陷型ii型cas9蛋白组合使用以将融合至抑制域的i型crisprcascade亚基蛋白募集和定位至基因靶标并抑制所述基因靶标的转录。
[0808]
实施例14
[0809]
i型cas基因的鉴定和筛选
[0810]
本实施例描述了从不同的物种鉴定和筛选i型cas基因的方法。这里提供的方法自shmakov,s.,et al.,mol.cell 60:385-397(2015)调整而来。
[0811]
a.i型crispr-cas基因的鉴定
[0812]
使用基本局部比对搜索工具(blast,blast.ncbi.nlm.nih.gov/blast.cgi),可以对各种物种的基因组进行搜索,以鉴定编码i型crispr-cas复合体的各种基因组分的一个或多个基因。cas1整合酶基因是第1类和第2类crispr-cas家族的组分,并且在鉴定出包含cas1基因的物种后,可以进行这些基因组中的子序列搜索,以分离出包含i型特异性基因的基因组。可以将基因组搜索锚定在crispr-cas整合酶基因cas1上,来自可以使用的来自大肠杆菌k-12mg1655的i-e型系统的示例性的cas1序列是seq id.no:621。特定基因(例如,cas7和cas5)是i型系统的干扰复合体的核心组分,并且可用于进一步区分包含i型系统的物种。可以使用的大肠杆菌k-12mg1655 cas7和cas5基因的示例性的序列分别是seq id.no:622和seq id.no:623。通过鉴定i型特异性核酸酶-解旋酶cas3基因或其同系物,可以进一步解析鉴定出的具有cas7和cas5基因的基因组。可以使用的大肠杆菌k-12mg1655 cas3序列的样本性的示例性的序列是seq id.no:624。
[0813]
包含crispr-cas整合酶基因cas1、i型干扰复合体基因cas7和cas5,以及核酸酶-解旋酶cas3基因,或以上的一些组合的基因组,可能是i型crispr-cas系统的候选物。i型crispr-cas基因通常被发现接近单个基因组位点中的一个,通常在20千碱基(kb)内。可以搜索cas1、cas7、cas5或cas3基因周围的区域,以寻找构成i型干扰复合体的其余cas基因的其他开放阅读框(orfs)。可以将推定的orfs的氨基酸序列与已知的i型基因进行同源性比较,或者可以使用通过max planck研究所生物信息学工具包(www.toolkit.tuebingen.mpg.de/#/)或等同物可获取的同源性检测和结构预测搜索工具分析i型蛋白组分的特征性蛋白域的存在。
[0814]
b.鉴定的i型组分的筛选
[0815]
一旦鉴定出i型成分(例如,cas基因和相应的crrna)的推定的集合,就可以测试i型成分进行可程序化dna靶向的能力。
[0816]
可以遵循实施例1、2和3的指导,将推定的cas基因和crrna编码至表达载体中。可以将编码各种cas基因和crrna的载体引入细菌菌株中,并按实施例4和5中所述的表达和纯化i型干扰复合体。可以经由sds-page凝胶分析来自尺寸排阻色谱(sec)柱的洗脱级分,以基于重量确定包含完整i型干扰复合体的蛋白质的性质。也可以运行溴化乙锭凝胶,以检测作为干扰复合体一部分的crrna的存在。
[0817]
如实施例6和7中所述,可以测试纯化的cascade复合体支持dna靶标的体外生化切割的能力。
[0818]
其中未表达单个推定的cas基因的对照表达和纯化样品可用于确定所需的cas基
因,这些cas基因构成能可编程的dna靶标的完整的i型干扰复合体。
[0819]
对于某些应用,从基因组序列中鉴定出单个cas基因同系物(例如,cas7)就足够了,并且不需要鉴定另外的cas基因或进行筛选。
[0820]
实施例15
[0821]
i型crrnas的鉴定
[0822]
本实施例描述了鉴定不同物种中的i型crrnas的方法。这里提供给的方法自chylinski,k.,et al.,rna biology 10:726-737(2013)调整而来。
[0823]
如实施例17a中所述,可以进行各种物种的基因组搜索以鉴定i型crispr-cas基因。包含一个或多个i型特异性cas基因的基因组是可能包含在crispr重复-间隔区阵列中编码的crisprrnas(crrnas)的候选基因组。可以探测与鉴定的i型cas基因(例如,cas7、cas5或cas3基因)相邻的序列,以寻找相关的crispr重复-间隔区阵列。可以遵循grissa,i.v.,et.al.nucleic acids res.35(网络服务器出版):w52-w57(2007),将用于计算机预测筛选的方法用于从重复阵列提取crrna序列。crrna序列包含在crispr重复阵列中,并且可以通过外来间隔区序列所间隔的其标志重复序列进行鉴定。
[0824]
a.rna-seq文库的制备
[0825]
可以使用rna测序(rna-seq)进一步验证包含在计算机中鉴定的单个crrna的推定crispr阵列。
[0826]
可以从商业资源库(例如,atcc,manassas,va;german collection of microorganisms and cell cultures gmbh(dsmz),braunschweig,germany)获得来自鉴定为包含推定的i型cas基因和crrna组分的物种的细胞。
[0827]
可以将细胞培养至对数中期,并使用trizol试剂(sigmaaldrich,st.louis,mo)准备总rna,并用dna酶i(fermentas,vilnius,lithuania)处理。
[0828]
可用ribo-zero rrna去除试剂盒(illumina,san diego,ca)处理10μg的总rna,并使用rna净化和浓缩器(zymo research,irvine,ca)纯化剩余的rna。
[0829]
可以按照制造商的指导,使用truseqtm小rna文库制备试剂盒(illumina,san diego,ca)来制备文库。这将导致具有适配体序列的cdnas。
[0830]
可以使用miseq测序仪(illumina,san diego,ca)对所得的cdna文库进行测序。
[0831]
b.测序数据的处理
[0832]
例如,可以使用以下方法来处理cdna文库的测序读段。
[0833]
可以使用cutadapt 1.1(pypi.python.org/pypi/cutadapt/1.1)去除适配体序列,并从读段的3'端修剪约15个核苷酸来提高读段质量。
[0834]
可以使用bowtie 2(www.bowtie-bio.sourceforge.net/bowtie2/index.shtml)将读段与各自物种(即待鉴定推定crrna的物种)的基因组对齐。可以使用samtools(www.samtools.sourceforge.net/)将通过bowtie 2生成的序列比对/图谱(sam)文件转换为二进制比对/图谱(bam)文件,以进行后续的测序分析步骤。
[0835]
可以使用bedtools(bedtools.readthedocs.org/en/latest/)从bam文件计算映射到crispr位点或基因座的读段覆盖。
[0836]
可以将上一步中生成的bed文件加载到综合基因组学查看器(igv;www.broadinstitute.org/igv/)中,以可视化测序读段堆积。读段堆可用于识别转录的推
定crrna序列的5'和3'端。rna-seq数据可用于验证推定的crrna元件在体内积极转录。
[0837]
可以遵循实施例17a的指导,用其同源i型cas基因测试推定的crrna进行可编程dna靶向的能力。
[0838]
实施例16
[0839]
耐受cascade向导rna骨架中的变化的位点的探测
[0840]
本实施例描述了i型向导crrnas的各种变化的产生和测试及其用于构建cascade多核苷酸复合体的适合性。以下描述的方法自briner,a.,et al.,mol.cell 56:333
–
339(2014)调整而来。
[0841]
可以将变化引入crrna骨架中,并利用同源的cascade复合体测试所得的改造的crrna,以促进适合改造的i型向导crrna骨架中的区域或位置的鉴定。
[0842]
可以选择来自i型crispr系统(例如,大肠杆菌cascade)的crrna进行改造。可以在计算机上改造crrna序列,以引入一个或多个碱基变化(例如,选自一个或多个以下区域的区域中的核酸序列中的取代、变化、突变、缺失和/或插入:间隔区的核酸序列5
’
(5
’
柄)、间隔区元件、i型crispr重复茎序列或i型crispr重复茎序列的3
’
(3
’
柄)。
[0843]
碱基变化也可用于在任何crrna区域的氢碱基对相互作用中引入错配,或通过取代两个碱基引入替代的氢碱基对相互作用的碱基对突变,其中替代的氢碱基对相互作用不同于原来的氢碱基对相互作用(例如,原来的氢碱基对相互作用是watson-crick碱基配对,且两个碱基的取代形成反向hoogsteen碱基配对)。碱基的取代也可以用于在crrna骨架内引入氢碱基对相互作用。
[0844]
crrna的区域可以独立地进行改造,以将二级结构元件引入crrna骨架中。这样的二级结构元件包括但不限于以下元件:茎-环元件、茎元件、假结和核糖酶。此外,可以对crrna骨架进行改造,以通过crrna的5'端、3'端或内部的缺失来删除crrna骨架的一部分。也可以引入替代的骨架结构。
[0845]
可以将计算机设计的crrna序列提供给商业制造商进行合成。
[0846]
可以评估改造的crrnas支持单个cascade亚基蛋白(即cas6、cas5等)结合的能力,或支持cascade蛋白复合体的完整形成的能力,或支持cascade复合体形成和通过募集核酸酶(例如,cas3)修饰双链dna靶序列的能力。可以通过纳米-esi质谱法以类似于jore,m.,et al.,nature structural&molecular biology 18:529
–
536(2011)的方式评估与单个cascade亚基蛋白和cascade蛋白复合体组装体结合的crrna。可以以与实施例6和7中所述的那些类似的方式,进行通过募集核酸酶的双链dna靶序列的crrna和cascade蛋白复合体修饰的生化定征。可以使用实施例8a、实施例8b、实施例8c和实施例8d中所述的方法,验证能够通过募集核酸酶支持cascade复合体的形成和双链dna靶序列的修饰的改造的crrna在细胞中的活性。
[0847]
实施例17
[0848]
包含dna靶标结合序列的cascade复合体向导的筛选
[0849]
本实施例示出了使用本发明的i型crispr蛋白和i型向导crrnas修饰人类gdna(gdna)中出现的dna靶序列,以及测量那些位点处的切割活性水平。
[0850]
可以首先从gdna选择靶标位点(dna靶序列)。i型向导crrnas可以设计为靶向选择的序列。可以进行测定(例如,如实施例7中所述的)以确dna靶序列切割的水平。
[0851]
a.从gdna选择dna靶序列
[0852]
可以鉴定选择的基因组区域中的cascade蛋白复合体(例如,大肠杆菌i-e型cascade)的pam序列(例如,atg)。
[0853]
可以鉴定3
’
临近atg pam序列的一个或多个cascade dna靶序列(例如,32个核苷酸长度的)。
[0854]
选择核酸靶序列的标准可以包括但不限于以下标准:与基因组中的其他区域同源;百分比g-c含量;溶化温度;在间隔区内存在均聚物;两条序列之间的距离;以及本领域技术人员已知的其他标准。
[0855]
可以将与cascade dna靶序列杂交的dna靶标结合序列掺入向导crrnas中。向导crrna构建体的核酸序列通常提供给商业制造商,并由其合成。
[0856]
可以将如本文所述的向导crrna与同源的i型cascade蛋白复合体一起使用,以形成crrna/cascade蛋白复合体。
[0857]
b.切割百分比和特异性的确定
[0858]
例如,可以使用实施例7中所述的切割测定确定与向导crrna相关的体外切割百分比和特异性(即脱靶结合的量),并按如下进行比较:
[0859]
(1)如果对于向导crrna只鉴定或选择了单个dna靶序列,则可以确定每个dna靶序列的切割百分比和特异性。如果需要的话,可以在进一步的实验中使用以下方法更改切割百分比和/或特异性,包括但不限于改造向导crrna,或引入效应子蛋白/效应子蛋白结合序列以改造向导crrna或cascade亚基蛋白,或配体/配体结合部分以改造向导crrna或cascade亚基蛋白。
[0860]
(2)如果对于向导crrnas鉴定了多种dna靶序列或选择,则可以比较从切割测定获得的包含靶标结合序列的不同dnas之间的切割百分比数据和位点特异性数据,以鉴定具有所需切割百分比和特异性的dna靶序列。切割比例数据和特异性数据提供了用于各种应用的基础选择的标准。例如,在一些情况下,向导crrna的活性可能是最重要的因素。在其他情况下,切割位点的特异性可能比切割比例相对更重要。如果需要的话,可以在进一步的实验中使用以下方法改变切割百分比和/或特异性,方法包括但不限于,改造向导crrna、引入效应子蛋白/效应子蛋白结合序列以改造向导crrna或cascade亚基蛋白,或配体/配体结合部分以改造向导crrna或cascade亚基蛋白。
[0861]
可选地,或除体外分析外,可以使用例如实施例8c和实施例8d中所述的方法获得向导crrnas的细胞内切割百分比和特异性,并按如下进行比较:
[0862]
(1)如果对于向导crrna只鉴定或选择了单个dna靶序列,则可以确定每个dna靶序列的切割百分比和特异性。如果需要的话,可以在进一步的实验中使用以下方法更改切割百分比和/或特异性,方法包括但不限于改造向导crrna,或引入效应子蛋白/效应子蛋白结合序列以改造向导crrna或cascade亚基蛋白,或配体/配体结合部分以改造向导crrna或cascade亚基蛋白。
[0863]
(2)如果对于向导crrnas鉴定或选择了多种dna靶序列,则可以进行从切割测定中获得的包含靶标结合序列的不同dnas的切割百分比数据和位点特异性数据的比较,以鉴定具有所需切割百分比和特异性的dna靶序列。切割百分比数据和特异性数据提供了用于各种应用的基础选择。例如,在一些情况下,向导crrna的活性可能是最重要的因素。在其他情
id no:825至seq id no:1016,而不是来自实施例8c的表36的引物y和z。
[0875]
d.深度测序数据分析
[0876]
基本上按照实施例8d中所述进行深度测序数据分析。图31a和图31b示出了数据分析的结果。在图31a和图31b中,基因组编辑百分比显示为foki-cas8连接子类型(图31a,图31b,垂直轴14-60aa)和间隔区间距(n=1)(图31a,图32b,水平轴,间隔区间距5-50bp的函数。在图31a中,右侧的灰度等级垂直条为插入缺失的百分比。在图32b中,单元格中的值为插入缺失的百分比。数据的初步分析显示17和20个氨基酸的(分别为seq id no:821和seq id no:822)且间隔区间距为~26bp和~30-32bp的foki-cas8连接子的基因组编辑最高。数据经过了重新处理,并去除了少于1000个序列读段的样品,因为由于覆盖率低它们可能包含虚高的编辑值(只有在所有相关的样品都包含>1000读段时才保留位点)。在图31a和图31b中提供的此数据表明17和20个氨基酸的(分别为seq id no:821和seq id no:822)且间隔区间距为~30-32bp的foki-cas8连接子的基因组编辑最高。因此,通过改变foki-cas8融合蛋白的间隔区间距和连接子多肽长度,实现了使用包含fok1-cas8融合蛋白的i型crispr-cas复合体的高效基因组编辑。本文讨论了连接子多肽的氨基酸组合物。
[0877]
实施例19
[0878]
鉴定用于基因组编辑的cascade同系物
[0879]
本实施例示出了多种同源cascade复合体的设计和测试以评估基因组编辑的效率。
[0880]
a.用于测试同源cascade复合体的位点的识别
[0881]
鉴定了一组位点来测试另外的同源cascade复合体。具体来说,最小crispr序列被设计为靶向人类基因组中具有30-bp间隔区间距且两侧为aag或atg pam序列的一组位点。按照实施例9a中关于seq id no:454所述的方法,克隆了含有“重复区-间隔区-重复区-间隔区-重复区”序列的向导多核苷酸。用于生成最小crispr阵列的全套寡核苷酸序列表示为seq id no:1017至seq id no:1130(hsa33f,seq id no:1017、和hsa33r,seq id no:1074,例如一对)。包括了包含靶向trac位点的向导的阳性对照(seq id no:454)。
[0882]
将foki-cascade rnp亚基蛋白质组分编码基因克隆到载体中,该载体包含:使得能够在哺乳动物细胞中进行递送和表达的cmv启动子;经由2a病毒肽“核糖体跳跃”序列连接的cas基因;包含与30-aa连接子(seqid no:455)连接的foki和cas8的融合蛋白。
[0883]
b.编码foki-cascade rnp复合体组分的载体的转染
[0884]
基本上按照实施例8b中所述进行转染条件,具有以下修改。进行核转染前,将5μl质粒载体溶液转移至96孔板的单个孔中。每个孔包含3μg的编码foki-cascade rnp亚基蛋白组分的质粒和0.3μg的编码最小crispr阵列的质粒。
[0885]
c.来自转染细胞的gdna的深度测序
[0886]
基本上按实施例8c所述进行了深度测序,具有以下修改。代替来自实施例8c的表36的引物y和z,用于本实施例的靶标特异性引物为seq id no:1131至seq id no:1244。
[0887]
d.深度测序数据分析
[0888]
基本上按照实施例8d中所述进行深度测序数据分析。图32示出了数据分析的结果。在图32中,除了来自实施例8a的靶标hsa07(n=3)外,还针对58个测试位点(图32,水平轴,“靶标”;用于产生这些最小crispr阵列的寡核苷酸序列在上文有论述)绘制了基因组编
辑百分比(图32,垂直轴,%编辑)。如图32所示,编辑的范围从~6%至低于检测限。从这些数据中,选择了一组8个位点(hsa07以及与具有aag pams的以下靶标hsa37、hsa43、hsa46、hsa60、hsa77、hsa88和hsa126相对应的靶标1、3-5、10、13和16)以测试同源cascade复合体的基因组编辑。
[0889]
e.用于测试foki核酸酶的基因组编辑的同源cascade复合体的识别
[0890]
来自不同i型系统的cas8蛋白序列被用作psi-blastp的查询,以产生同系物选择的系统发育树。具体地,来自具核梭杆菌(wp_008798978.1)的cas8被用于i-b型,来自嗜碱耐盐芽孢杆菌(wp_010896519.1)的cas8被用于i-c型,来自大肠杆菌(wp_001050401.1)的cas8被用于i-e型,来自铜绿假单胞菌(wp_003139224.1)的cas8被用于i-f型,并且来自腐败希瓦氏菌(wp_011919226.1)的cas5被用于i-fv2型。
[0891]
接下来,对psi-blastp进行多次迭代,直到为每种i型系统识别出数千个同系物。从该信息,使用生命交互树(the interactive tree of life)在线软件(itol,accessible at itol.embl.de/login.cgi)构建了系统发育树。在使用可变分支长度自动折叠分支后,目视检查树。
[0892]
然后将落入主要进化枝中的生物体列表输出并手动检查以进行选择。在此步骤中,对于i-e型中的12种同系物以及i-b、i-c、i-f和i-fv2型的2-3种代表性的同系物,优先考虑选择从系统发育树的不同区域采样的同系物。基于上述系统发育分析,将cas8和cas5候选序列输入到ncbi中,并在ncbi的基因组图形浏览器中目视检查内源宿主细菌内的基因组内容物。只有在以下条件下才选择cascade同系物:(1)它们在37℃下生长的生物体中被发现;(2)它们的cas基因操纵子是完整的,并具有所有预期的cascade亚基蛋白编码基因、cas3基因和完整的获得基因(即cas1和cas2);(3)它们的cas基因操纵子两侧是一个或多个crispr序列;以及(4)它们的crispr阵列包含>10个间隔区。对于一些同系物,使用crisprfinder程序(crispr.i2bc.paris-saclay.fr/server/)来识别推定的pam序列。根据上述标准,选择表44所示的22种同源cascade复合体。
[0893]
[0894][0895]
*如sinkunas,t.,et al.,embo j.32:385-394(2013)所鉴定的;然而,本文示出的数据证明铜绿假单胞菌菌株nd07可以在体内利用单个a作为pam序列。
[0896]
f.用于转染至靶细胞中的编码来自22种不同的物种的foki-cascade rnp组分的载体的产生
[0897]
来自每种同系物的每个cas基因的序列被合成为多顺反子构建体的一部分,该构建体包括包含foki核酸酶和cas8的融合蛋白。对于每种i-e型cascade复合体同系物,生成了一组~7-8个靶向带有适当pam序列的位点的向导。对于每种i-b、i-c、i-f和i-fv2型cascade同系物,生成了一组~2-7个靶向带有适当pam序列的位点的向导。每种cascade复合体同系物系统都需要独特的重复序列来处理其同源向导(seq id no:1267至seq id no:1288)。使用实施例9a中关于seq id no:454所述的方法,克隆包含“重复区-间隔区-重复区-间隔区-重复区”序列的向导的编码序列。寡核苷酸在5'端被磷酸化,并附接有突出序列,从而能够克隆到具有适当重复序列的质粒载体中。用于产生22种cascade复合体同系物的最小crispr阵列的全套寡核苷酸序列表示为(seq id no:1289至seq id no:1400)。
[0898]
将foki-cascade rnp亚基蛋白组分编码基因克隆到载体中,该载体包含:使得能够在哺乳动物细胞中进行递送和表达的cmv启动子;经由2a病毒肽“核糖体跳跃”序列连接的cas基因;包含利用30-aa连接子连接的foki和cas8的融合蛋白。
[0899]
g.编码foki-cascade rnp复合体的质粒的转染
[0900]
基本上按实施例8b中所述进行转染条件,具有以下修改。进行核转染前,将5μl质粒载体溶液转移至96孔板的单个孔中。每个孔中包含1.5μg的编码foki-cascade rnp亚基蛋白组分的质粒和~0.5-1.5μg的编码最小crispr阵列的质粒。实验以一式三份进行,并且包括来自大肠杆菌的靶向8个位点(来自实施例8a的hsa07以及来自实施例19f和实施例19g的hsa37、hsa43、hsa46、hsa60、hsa77、hsa88、hsa126)的foki-cascade rnp复合体(seq id no:455)作为阳性对照。如先前所述,以下寡核苷酸被用于产生与大肠杆菌阳性对照一起使用的最小crispr阵列:hsa37(seq id no:1019;seq id no:1076)、hsa43(seq id no:1024;
seq id no:1081)、hsa46(seq id no:1027;seq id no:1084)、hsa60(seq id no:1037;seq id no:1094)、hsa77(seq id no:1045;seq id no:1102)、hsa88(seq id no:1050;seq id no:1107)、hsa126(seq id no:1072;seq id no:1129)。
[0901]
h.来自转染细胞的gdna的深度测序
[0902]
基本上按实施例8c所述进行了深度测序,具有以下修改。代替来自实施例8c的表36的引物y和z,用于本实施例的靶标特异性引物为seq id no:1401至seq id no:1512。对于两种i-e型rnp复合体和i-b、i-c、i-f和i-fv2型rnp复合体,包括了包含大肠杆菌i-e型cascade的对照样品,以便进行比较,并利用与来自实施例8a的靶标hsa07以及来自本实施例的hsa37、hsa43、hsa46、hsa60、hsa77、hsa88、hsa126相对应的靶标特异性引物进行测序。更具体地,对于这些靶标使用了以下靶标特异性的扩增引物:hsa37(seq id no:1133;seq id no:1190)、hsa43(seq id no:1138;seq id no:1195)、hsa46(seq id no:1141;seq id no:1198)、hsa60(seq id no:1151;seq id no:1208)、hsa77(seq id no:1159;seq id no:1216)、hsa88(seq id no:1164;seq id no:1221)、hsa126(seq id no:1186;seq id no:1243)。
[0903]
i.深度测序数据分析
[0904]
基本上按实施例8d所述进行深度测序数据分析。图33a和图33b显示了来自这些实验的结果。在图33a中,垂直轴是编辑百分比(图33a,%编辑),并且水平轴上的数字是与i-e型同系物系统相对应的seq id编号。利用许多i-e型foki-cascade同系物观察到编辑(图33a)。利用来自假单胞菌s-6-2的变体观察到最高编辑,而其他同系物(即肠道沙门氏菌、地热杆菌epr-m、稻田甲烷胞菌mre50和嗜热链球菌(菌株nd07))显示大约等于大肠杆菌的编辑。在图33b中,垂直轴是编辑百分比(图33b,%编辑),并且水平轴上的数字是与i-b、i-c、i-f和i-fv2型同系物系统相对应的seq id编号。利用来源于i-b、i-c、i-f和i-fv2型的foki-cascade rnps的编辑低于检测限(图33b)。
[0905]
本实施例提供了筛选i型同系物以鉴定提供基因组编辑能力的i型系统的方法。另外的i型同系物筛选描述于实施例22。
[0906]
实施例20
[0907]
假单胞菌s-6-2中用于高效基因组编辑的不同foki-cas8连接子长度和间隔区间距
[0908]
本实施例示出了包含不同长度的foki-cas8和连接子多肽的多种融合蛋白的设计和测试,以及不同间隔区间距对利用假单胞菌s-6-2i-e型crispr-cas系统的高效基因组编辑的影响。
[0909]
a.编码待转染至靶细胞中的foki-cascade rnp组分的载体的产生
[0910]
最小crispr阵列被设计为靶向人类基因组中的一组位点。间隔区间距范围为23-34bp,增量为1bp。每个间隔区间距设计了八种靶标,并且靶标的两侧是aag pam序列。使用3种寡核苷酸(seq id no:1513至seq id no:1515)和编码使得能够进行foki-cascade靶向的“重复区-间隔区-重复区-间隔区-重复区”序列的独特的引物,利用基于pcr的组装(寡聚物模板化的pcr扩增)产生了最小crispr阵列。用于产生最小crispr阵列的整套独特的寡核苷酸序列是seq id no:1516至seq id no:1704。基本上按照制造商的说明,使用(beckman coulter,pasadena,ca)珠子纯化和浓缩pcr组装的向导。
[0911]
将foki-cascade rnp亚基蛋白组分编码基因克隆至载体中,该载体包含:使得能够在哺乳动物细胞进行递送和表达的cmv启动子、经由2a“核糖体跳跃”序列连接的cas基因和利用30-aa连接子(seq id no:1748)附接至cas8的foki。设计了不同长度的另外的连接子多肽序列,并用于将foki与cas8蛋白连接以形成融合蛋白。连接子多肽序列列出在表45中。
[0912][0913]
b.编码foki-cascade rnp复合体组分的载体的转染
[0914]
除了具有以下修改外,基本上按照实施例8b中所述进行转染条件。进行核转染前,将5μl质粒载体溶液转移至96孔板的单个孔中。每个孔包含5μg编码foki-cascade rnp蛋白组分的质粒和~0.1-0.5μg编码最小crispr阵列的线性pcr产物。
[0915]
c.来自转染细胞的gdna的深度测序
[0916]
基本上按实施例8c中所述进行深度测序。靶标特异性引物是seq id no:1705至seq id no:1803,而不是来自实施例8c的表36的引物y和z。
[0917]
基本上按实施例8d中所述进行深度测序数据分析。图34显示了95个位点(n=1)处的基因组编辑(图34,垂直轴“%编辑)。在图34中,水平轴对应于碱基对的间隔区间长度(图34,bp间隔区间)。从左至右的3幅条形图17aa(图34,空心条)、20aa(图34,交叉线条)和30aa(图34,条形条)代表的连接子长度。编辑范围为~50%(图34,误差条,显示了均值+/-1s.d.)至低于检测限,并且与间隔区间距和连接子多肽长度相关。本文中讨论了连接子多肽的氨基酸组合物。~30-33bp的间隔区间距和17和20个氨基酸的连接子多肽长度提供了非常高效的编辑。
[0918]
支持本发明的来自基本上根据本实施例中所示的相同的方案进行的另外的实验的数据提供在图41a、图41b和图41c中。在这些图中,垂直轴是编辑效率(%),并且水平轴是bp的间隔区间距(23-34bp)。数据扩充了3种cascade同系物变体foki-psecascade(图41a)、foki-ecocascade(图41b)和foki-sthcascade(图41c)的foki-cas8连接子长度和间隔区间距的筛选。编辑效率百分比表示为17aa、20aa和30aa(图41a、图41b和图41c:从左至右,17aa、20aa和30aa)和间隔区间距的foki-cas8连接子长度的函数。每个点代表单个基因组位点,并且每个间隔区间距测试7
–
8个位点。均值显示在条形图中。如从这些数据可以看出的,~30-33bp的间隔区间距和17、20和30个氨基酸的连接子多肽长度为foki-psecascade提供了高效的编辑,~31-33bp的间隔区间距和17、20和30个氨基酸的连接子多肽长度为foki-ecocascade提供了高效的编辑,并且~29-31bp的间隔区间距和17、20和30个氨基酸的连接子多肽长度为foki-sthcascade提供了高效的编辑。
[0919]
实施例21
[0920]
利用cas3-foki和foki-cas8使得能够进行foki-cascade基因组编辑
[0921]
本实施例示出了使用cas3-foki和foki-cascade来诱导foki的二聚化,以在人类基因组中的位点处产生双链断裂(参见,例如图16a、图16b和图16c)。更具体地,本实施例详
细描述了用于产生基因组编辑效率的多种cas3-foki连接子组合物和长度以及foki-cas8连接子组合物和长度的设计和测试。
[0922]
a.编码待转染至靶细胞中的foki-cas3和foki-cascade rnp组分的载体的产生
[0923]
最小crispr阵列被设计靶向人类基因组中的两侧为aag pams的三个不同的位点。选择的位点以前显示支持通过向导指导的大肠杆菌foki-cascade二聚物的间隔区间编辑,并因此已知可以允许foki-cascade结合(例如,hsa37、hsa43和hsa46)。
[0924]
上面实施例中描述的foki-cascade系统使用了两种foki cascade复合体(参见,例如图15a、图15b和图15c);因此,可以使用指定第一核酸靶标位点的第一向导序列和指定第二核酸靶标位点的第二向导序列。由于cas3-foki-foki-cascade系统仅需要单个pam,因此包含“重复区-间隔区-重复区”的向导应足以促进功能cascade复合体与核酸靶标位点的结合。也可以使用含有“重复区-间隔区-重复区-间隔区-重复区”的多核苷酸,但是通常在本实施方案中,两个间隔区序列指导cascade复合体结合到相同的核酸靶序列上;也就是说,两个间隔区可以具有相同的序列。基本上按实施例9a中关于seq id no:454所述克隆向导。以下退火的寡核苷酸被用于产生最小crispr阵列:hsa37(seq id no:1019;seq id no:1076)、hsa43(seq id no:1024;seq id no:1081)和hsa46(seq id no:1027;seq id no:1084)。
[0925]
如实施例9a中所述,将foki-cascade rnp蛋白组分编码基因克隆至含有mv启动子的质粒载体中,以能够使得在哺乳动物细胞中进行递送和表达。cas基因经由2a“核糖体跳跃”序列连接。此外,利用30-aa连接子(seq id no:455)将foki融合至cas8。设计了不同长度的另外的连接子序列和组合物,并用于连接foki至cas8蛋白。这样的序列的实例列出在表46中。
[0926]
使用30-aa连接子在c端将来自大肠杆菌的cas3蛋白与foki融合。利用nls序列在n端(seq id no:1806)进一步改造该融合物。设计了不同长度的另外的连接子序列和组合物,并用于将foki连接至cas3蛋白(表46和seq id no:1804至seq id no:1807)。
[0927]
产生了另外的cas3-foki融合构建体,其中cas3蛋白的解旋酶或核酸酶活性被失活(seq id no:1808至seq id no:1815)。解旋酶和核酸酶活性通过分别制造cas3蛋白的d452a和d75a突变而受损(参见,例如mulepati,s.,et al.,j.biol.chem.288:22184-22192(2013))。
[0928][0929]
b.编码foki-cascade rnp复合体的质粒的转染
[0930]
按实施例8b所述进行转染条件,具有以下修改。进行核转染前,将5μl质粒载体溶液转移至96孔板的单个孔中。每个孔包含以下三种组分:3μg的编码一组foki-cascade rnp
蛋白组分的质粒、3μg的编码cas3-foki的质粒和0.5μg的编码最小crispr阵列的质粒。将96孔板设置为基质,以提供三种组分的所有组合。
[0931]
c.来自转染细胞的gdna的深度测序
[0932]
按实施例8c中所述进行深度测序,具有以下修改。代替来自实施例8c的表36的引物y和z,本实施例中使用的靶标特异性引物如下:seq id no:1133和seq id no:1190(hsa37靶标位点)、seq id no:1138和seq id no:1195(hsa43靶标位点),以及seq id no:1141和seq id no:1198(hsa46靶标位点)。
[0933]
d.深度测序数据分析
[0934]
按实施例8d中所述进行深度测序数据分析,除了记录foki-cascade结合位点pam序列上游的~1bp至~25bp的插入缺失外。以这种方式,可以确定支持最有效编辑的foki-cas8连接子序列、cas3-foki连接子序列和cas3变体的组合。
[0935]
实施例22
[0936]
筛选改造的同源foki-cascade复合体
[0937]
本实施例示出了具有不同数量的亚基的多个同源的cascade复合体的设计和测试,以评估基因组编辑的效率。本实施例扩展了实施例19中描述的分析。
[0938]
a.待转染至foki-cascade rnp复合体的靶细胞中的dna模板组分的产生
[0939]
设计了最小crispr阵列,以将两种foki-cascade rnp复合体靶向人类基因组中gdna的相对链上的相邻位点。foki-cascade构建体来源于含有以下3种或4种基因的11种同源的物种中的每种:具核梭杆菌(fnu,i-b型)、胎儿弯杆菌(cfe,i-b型)、内脏臭气杆菌(osp,i-b型)、嗜碱耐盐芽孢杆菌(bhe,i-c型)、普通脱硫弧菌(dvu,i-c型)、霍乱弧菌菌株l15(vch,i-f型)、产酸克雷伯氏菌(koh,i-f型)、铜绿假单胞菌(pae,i-f型)、腐败希瓦氏菌(spu,i-fv2)、不动杆菌(aci,i-fv2型)、霍乱弧菌菌株he48(vch_v2,i-fv2型)。
[0940]
设计了第一和第二改造的第一类i型crispr-cas效应子复合体,其中第一向导多核苷酸包含能够结合第一核酸靶序列的第一间隔区,第二向导多核苷酸包含能够结合第二核酸靶序列的第二间隔区,并且第一核酸靶序列的pam和第二核酸靶序列的pam具有14个碱基对至60个碱基对的间隔区间距。两种改造的第一类i型crispr-cas效应子复合体的定向使得相对于向导rna靶序列,pams朝向内(即,pam在内的方向)。pam序列对于i-b型是tca,对于i-c型是ttc,并且对于i-f、i-fv2型(在crispr阵列中,i-f型和i-fv2型具有不同的重复序列;参见表47和表44)是cc。
[0941][0942]
基本上按本文所述(例如,实施例20a;以及图42a和图42b),使用3种寡核苷酸和编码使得能够进行foki-cascade rnp复合体靶向的“重复区-间隔区-重复区-间隔区-重复区”序列的独特的引物,利用基于pcr的寡聚物模板化的组装体产生了最小crispr阵列。对于i-b型和1-c型,使用了非通用的反向寡核苷酸引物。基本上按实施例20a中所述,使用
珠子(beckman coulter,pasadena,ca)纯化和浓缩pcr组装的最小crispr阵列。
[0943]
在改造的第一类i型crispr-cas效应子复合体中,对于i-b型、i-c、i-f复合体,将foki编码序列融合至cas8的n端,且对于i-fv2型复合体,融合至cas5的n端。将foki-cascade rnp蛋白组分编码基因克隆至包含以下成分的载体(参见表44和表47)中:使得能够在哺乳动物细胞中递送和表达的cmv启动子、经由2a“核糖体跳跃”序列连接的cas基因,以及利用30-aa连接子附接至cas8(或在i-fv2型同系物的情况下利用30-aa连接子附接至cas5)的foki单体。
[0944]
b.编码改造的foki-cascade rnp复合体组分的载体的转染
[0945]
基本上按照实施例8b中所述进行转染条件,具有以下修改。进行核转染之前,将5μl的含有dna模板的溶液转移至96孔板的单个孔中,其中孔中含有约1.5μg的编码同源的foki-cascade复合体的组分的每种质粒,以及0.4μg的编码最小crispr阵列的线性pcr产物。
[0946]
c.来自转染的细胞的gdna的深度测序
[0947]
基本上按实施例8c中所述进行深度测序。然而,代替来自实施例8c的表36的引物y和z,使用了不同的靶标特异性引物。图43示出了数据分析的结果。在图43中,基因组编辑百分比显示为foki-cascade同系物变体的函数(图43,水平轴,11种同系物变体由以上所示的缩写标识,并且在水平轴上以相同的顺序出现)和间隔区间距(图43,垂直轴,14-60bp);右侧的灰度等级垂直条为插入缺失的百分比。以给定间隔区间距的每次测量均代表4个靶标位点间的平均编辑(每个靶标位点n=1)。在测试的靶标位点中,大部分改造的foki-cascade直系同源复合物的编辑均低于检出限,而使用改造的霍乱弧菌菌株l15(i-f型)foki-cascade复合体编辑的范围从低于检出限至高达~2%插入缺失,其中间隔区间距为26bp至28bp时观察到最高编辑。foki-cascade复合体在改造后的霍乱弧菌菌落he48(i-fv2型)中也观察到,其范围从检测限以下到1.5%,间隔区之间在42bp和46bp之间。利用改造的霍乱弧菌菌株he48(i-fv2型)foki-cascade复合体也观察到编辑,范围为低于检测限至~1.5%,间隔区间距为42bp至46bp。
[0948]
本实施例中的数据说明了本文详细描述的方法可以有效地用于鉴定能有效进行基因组编辑的同源的cascade复合体。
[0949]
实施例23
[0950]
使用mcas3蛋白限制细胞中的缺失长度
[0951]
本实施例示出了如何突变cas3蛋白,使得所得的cas3诱导的缺失相对于利用wtcas3蛋白产生的那些更短,以用于基因组编辑(例如,在人类细胞中)。
[0952]
a.cascade和cas3 dna模板组分的产生
[0953]
设计了最小crispr阵列以将大肠杆菌cascade(ecocascade)rnp复合体靶向在人类基因组中的chr2(hzgj基因)上具有aag pam的基因组位点。接下来,利用基于pcr的组装,使用3种寡核苷酸(seq id no:1513至seq id no:1515;实施例20a)和编码使得能够进行ecocascade rnp靶向(seq id no:1818)的“重复区-间隔区-重复区-间隔区-重复区”序列的独特的引物产生了最小crispr阵列。所得的扩增子包含驱动最小crispr阵列的表达的hu6启动子。对于该最小crispr阵列,将相同的序列用于两种间隔区序列。使用
珠子(beckman coulter,pasadena,ca)纯化和浓缩pcr组装的最小cripsr阵列。
[0954]
为了降低突变体蛋白质在dna上的dna易位性(即沿着dna的长度移动),设计了一系列的cascas3(ecocas3)突变体变体。设计了一组大肠杆菌cas3(ecocas3)突变体变体,以降低突变体蛋白在dna上的dna易位进行性(即沿dna的长度移动),并使保持dna核酸酶活性。
[0955]
参考结合至单链dna底物(huo,y.,et.al.,nat.struct.mol.biol.(9):771-777(2014))的褐色嗜热裂孢菌cas3的晶体结构、功能蛋白域的位置和与其他cas3直系同源物的同源,在ecocas3(大肠杆菌(p38036)cas3氨基酸序列:uniprotkb-p38036(cas3_ecoli))中制作了一组24种不同的突变,以调节解旋酶域中的atp结合/水解区域(即g317a、s318a、g319a、k320n、t321n、q297e、d452e、e453n、r662a、r665q)或解旋酶域的ssdna环结合/ssdna路径保守区域(即t346a、q347n、g375a、k412g、t423a、d425h、q426t、h601a、a602v、r603q、r609s、t635a、q636a、q640h)。表48列出了ecocas3野生型蛋白和突变体蛋白、编码序列(核苷酸序列)的质粒和相应的氨基酸序列。
[0956][0957]
*相对于野生型ecocas3蛋白序列
[0958]
将ecocascade rnp蛋白组分编码基因以及野生型(wt)和突变体ecocas3基因克隆至包含cmv启动子的载体中,以使得能够在哺乳动物细胞中进行递送和表达。经由2a“核糖
体跳跃”序列连接ecocascade rnp cas基因,并且所有基因都包含n端nls序列以将编码的蛋白导向核(ecocascade多顺反子质粒,核苷酸序列seq id no:1871,多顺反子的氨基酸序列1872)。
[0959]
b.编码改造的ecocascade rnp的载体、野生型ecocas3蛋白和突变体ecocas3蛋白的转染
[0960]
基本上按实施例8b中所述进行转染条件,具有以下修改:进行核转染之前,将6μl的包含dna模板的溶液转移至96孔板的单个孔中
–
含有3μg的编码ecocascade复合体蛋白的质粒、1μg的编码野生型或突变体ecocas3蛋白的质粒以及0.2μg的编码最小crispr阵列的线性pcr产物的孔。在转染后约4天收获gdna。
[0961]
c.来自转染的细胞的gdna的深度测序
[0962]
基本上按实施例8c中所述进行深度测序。然而,靶标特异性引物是seq id no:1873至seq id no:1874,而不是来自实施例8c的表36的引物y和z;而且,使用了miseq试剂盒v3,600个循环(illumina,san diego,ca)。基本上按照实施例8d中所述进行深度测序数据分析,具有以下修改:(1)记录了在扩增子(扩增子位置:chr2:68156987-68157510;长度=524个核苷酸)窗中的任何位置具有至少一个读段且具有大于3个核苷酸的缺失的独特读段类别(在本文中称为“独特的缺失类别”;类别不会按读段计数加权,因为扩展偏差可能会影响具有较长缺失的产品的读段计数),(2)具有插入或多个缺失的读段类别被丢弃,和(3)缺失起始位点和终止位点被映射在样本之间比较。
[0963]
图45a、图45b、图45c和图45d显示了利用包含野生型ecocas3蛋白(n=21)、缺少ecocas3蛋白(n=3)或突变体ecocas3蛋白(n=3)的ecocascade rnp复合体在hzgj位点处的基因组编辑。图45a在垂直轴上显示了独特缺失类别的数量(图45a,0-600),并且在水平轴上显示了ecocas3蛋白变体(图45a,从左至右,野生型对照(wt)、无cas3蛋白对照和m1cas3蛋白至m24cas3蛋白,以表48中给出的顺序)。这里,引起524bp扩增子窗中的独特缺失类别数量增加的cas3突变体变体是用于减少易位进行性(即沿dna长度移动)的候选物。图45b在垂直轴上显示了碱基对的平均缺失长度,并且在水平轴上显示了ecocas3蛋白变体(关于图45a的相同的顺序)。与独特的缺失类别测量一样,在524bp扩增子窗口内产生较小缺失长度的cas3突变体变体是用于减少易位进行性的候选物。图45c在垂直轴上显示了相对于ecocascade pam上游6bp位点(即cas3切口位点附近)的平均缺失起始位置(bp),并且在水平轴上显示了ecocas3蛋白变体(关于图45a的相同顺序)。图45d在垂直轴上显示了相对于ecocascade pam上游6bp位点(即预期的cas3切口位点附近)的平均缺失终止位置(bp),并且在水平轴上显示了ecocas3蛋白变体(关于图45a的相同顺序)。这里,显示缺失起始和终止位置更接近ecocas3预期的切口位点的cas3突变体,被认为是用于减少易位进行性(即沿着dna长度移动)的强候选物。总之,显示出扩增子窗中增加的独特缺失类别、扩增子窗中的较短的缺失类别以及扩增子窗中的位置移位的缺失类别的一些组合的cas3突变体,是用于减少易位进行性的强候选物。
[0964]
数种突变体给出了指示减少了缺失长度的改变的修复模式。相对于野生型ecocas3蛋白,突变体ecocas3蛋白d452h和a602v均显示:(1)扩增子窗内独特缺失类别的数量大量增加,其可以指示更短的缺失,和(2)在扩增子窗中,缺失移到了相对于野生型ecocas3蛋白更接近ecocas3起始位点,其也可以指示更短的缺失。突变体ecocas3蛋白
a602v在扩增子窗中相对于野生型ecocas3蛋白也显示出更小的缺失。突变d452h和a602v均已预测会影响ssdna环结合。本实施例中的数据表明,与cascade rnp复合体结合时,当引入和在人类细胞中表达时,可以在cas3蛋白中引入突变以减少相对于wtcas3蛋白的缺失长度,并提供有关如何制备和使用包含突变的cas3蛋白来调节细胞中gdna的缺失长度的指导。
[0965]
实施例24
[0966]
使用路障限制cas3诱导的缺失长度
[0967]
在本申请中描述了数种限制和/或限定通过与cas3蛋白相关的cascade rnp复合体所促进的缺失长度的方法。这就是如何使用蛋白质包版限制cas3缺失的原因。本实施例示出了如何能够将蛋白路障用于限制cas3缺失。
[0968]
a.cas3蛋白和ecocascade rnp dna模板组分的产生
[0969]
设计了最小crispr阵列,以将大肠杆菌cascade(ecocascade)rnp靶向人类基因组中chr2(hzgj基因)上具有aag pam的基因组位点。接下来,基本上按实施例20a中所述,使用寡核苷酸(seq id no:1513至seq id no:1515)和编码使得能够进行ecocascade rnp靶向的“重复区-间隔区-重复区-间隔区-重复区”序列的引物,利用基于pcr的组装产生了最小crispr阵列。对于该最小crispr阵列,两个间隔区序列是相同的。主要根据制造商的说明,使用(beckman coulter,pasadena,ca)珠子纯化和浓缩pcr组装的向导。将改造的ecocascade蛋白组分编码基因以及大肠杆菌cas3(ecocas3)基因克隆至含有cmv启动子的载体中,以使得能够在哺乳动物细胞中进行递送和表达。经由2a“核糖体跳跃”序列(质粒核苷酸序列,seq id no:1871;多顺反子的蛋白序列,seq id no:1872)连接ecocascade rnp cas基因,并且所有基因均包含n端nls序列以将编码的蛋白导向核。
[0970]
b.dcas9-vp64/sgrna rnp复合体的产生
[0971]
其中复合体被用作路障以终止与cascade rnp复合体相关的cas3蛋白的易位进行性(即沿dna移动)的dcas9-vp64/sgrna rnp复合体的sgrna组分,通过体外转录(t7快速高产率rna合成试剂盒,new england biolabs,ipswich,ma)而产生。使用5'重叠引物的pcr被用于组装dsdna模板,以用于sgrna组分的转录。dsdna模板在dna序列的5'端处掺入了t7启动子。表49中示出了用于产生sgrna模板的组分、模板和引物。
[0972][0973]
组装sgrna dna模板的pcr反应按如下进行,其中反应混合物包含:一种浓度为40nm的“内部”dna引物(seq id no:1889至seq id no:1899),两种浓度为500nm的“外部”dna引物引物(seq id no:1887和seq id no:1888;包含t7启动子和rna序列的3'端)。基本上按照制造商的说明,使用q5热启动高保真2x预混液(new england biolabs,ipswich,ma)进行pcr反应。使用以下热循环条件进行pcr组装反应:98℃ 2分钟、11个循环的98℃ 10秒、58℃ 20秒、72℃ 20秒,以及最终的72℃下延伸1分钟。
[0974]
在37℃下,使用t7高产率rna合成试剂盒(new england biolabs,ipswich,ma),将约0.25-0.5μg的每种sgrna dna模板转录约16小时。转录反应物经dna酶i处理(new england biolabs,ipswich,ma)。具有融合至c端的vp64效应子域并且nls标签附接在vp64的c端上的dcas9蛋白(d10a&h840a;参见,例如sander,j.d.,et al.,nat.biotechnol.32:347
–
355(2014)),(n-nls-vp64编码序列-dcas9编码序列-c),从大肠杆菌中的细菌表达载
体(bl21(de3))表达而来,并使用亲和色谱、离子交换色谱(iec)和尺寸排阻色谱(sec),基本上按jinek,m.,et al.,science 337:816-821(2012)所述进行纯化。
[0975]
c.编码ecocas3的载体和ecocascade rnp复合体组分的组分以及dcas9-vp64/sgrna rnp复合体的转染
[0976]
基本上按照实施例8b所述进行hek293细胞的转染,具有以下修改:
[0977]
对于cas3/ecocascade rnp复合体形成,将4μl的包含编码ecocas3蛋白和ecocascade蛋白的dna模板的溶液转移至96孔板的单个孔中,其中所述孔包含3μg的编码ecocascade蛋白的质粒、0.2μg的编码最小crispr阵列的线性pcr产物,以及0、1或3μg的编码ecocas3的质粒;以及
[0978]
对于cas3-ecocascade rnp复合体形成,3μg的编码cas3-ecocascade蛋白组分的质粒,其中cas3利用17-aa连接子连接至cas8蛋白,并且0.2μg的编码最小crispr阵列的线性pcr产物。
[0979]
接下来,组装了dcas9-vp64/sgrna rnp复合体。具体地,将sgrnas在95℃下孵育2分钟,然后允许其平衡至室温约5分钟。将dcas9-vp64蛋白与sgrna一起在反应缓冲液(20mm hepes,ph 7.5,100mm kcl,5mm mgcl2,5%甘油)中以1:3的比例于37℃下混合10分钟。将组装的dcas9-vp64/sgrna rnp复合体以不同的剂量转移到96孔板的孔中,以转染到细胞中,建立基质,其中每种cas3/ecocascade或cas3-ecocascade混合物接受0、5、20或50pmol的dcas9-vp64路障。核转染后4天从细胞中收获gdna。
[0980]
d.来自转染的细胞的gdna的深度测序
[0981]
基本上按照实施例23c中的描述进行深度测序和数据分析。图46a、图46b和图46c示出了一系列的热图,这些热图表明了在dcas9-vp64/sgrna rnp复合体路障缺失或存在的情况下,cas3/ecocascade(利用1μg或3μg的cas3表达质粒,分别为图46a和图46b)或cas3-ecocascade(图46c)的与所示的dcas9-vp64/sgrna rnp复合体所靶向结合的位置(即,路障的位置,图46a、图46b、图46c,黑色箭头)相关的hzgj位点(图46a、图46b和图46c,空心箭头表示预期的cas3切口位点)处的缺失起始位点的频率。在hzgj位点处总共评估了11个路障(f1-f6和r1-r5)。在图46a、图46b和图46c中,“f”是指dcas9-vp64/sgrna rnp复合体的正向取向,其中正向取向意指与朝向ecocascade rnp复合体的核酸靶标结合位点的pam的dcas9-vp64/sgrna rnp复合体的核酸靶标结合位点相关的pam;“r”是指dcas9-vp64/sgrna rnp复合体的反向取向,其中反向取向意指与dcas9-vp64/sgrna rnp复合体的核酸靶标结合位点相关的pam不朝向ecocascade rnp复合体的核酸靶标结合位点的pam。在靶标位点指示器(f1-f6和r1-r5)的右侧,数字1、2、3和4分别对应于0、5、20或50pmol的dcas9-vp64/sgrna rnp复合体。每张热图上方的数字(-440至+100)对应于扩增子窗中的bp,其中0位点被指定为ecocascade rnp pam上游6bp。每张热图左侧的灰度等级条代表突变体类别的分数(0.0-0.5)。对于路障f4、f5和f6,缺失起始位点似乎高度富集在dcas9-vp64/sgrna rnp复合体路障放置位点附近。
[0982]
图47a和图47b显示了用3μg cas3-ecocascade和0pmol(图47a)或50pmol(图47b)dcas9-vp64/sgrna rnp复合体路障进行核转染的样品的扩增子窗中所有缺失的数据。在图47a和图47b中,空心箭头指示ecocas3蛋白切口位点的相对位置。在图47b中,黑色箭头显示了路障设置;即dcas9-vp64/sgrna rnp复合体的靶标结合位点)。在图47a和图47b中,垂直
轴代表缺失的3'端,单位为在扩增子窗内的bp,并且“0”位点是ecocascade rnp pam上游指定为6bp的位点;并且水平轴代表缺失的5'端,单位为在扩增子窗内的bp,并且“0”位点被指定为ecocascade rnp pam上游6bp。在图47a和图47b中,水平虚线代表缺失的3
’
端的平均位置,并且垂直虚线代表缺失的5
’
端的平均位置。图47a和47b中的每个的顶部上的条形图对应于缺失的5'端的分布,并且曲线代表缺失的5'端的核密度估计。类似地,图47a和47b中的每个右边的条形图对应于缺失的3'端的分布,并且曲线代表缺失的3'端的核密度估计。缺失起始位点在图47b中的黑色箭头附近高度富集,强烈表明路障阻止了cas3删除路障上游的gdna。
[0983]
本实施例中的数据支持使用蛋白质路障来控制由与cascade rnp复合体相关的cas3蛋白介导的缺失长度;因此,提供了使用与cascade rnp复合体相关的cas3蛋白来促进细胞的gdna中的具有限定长度的缺失的形成的方法。
[0984]
实施例25
[0985]
使用连接至cascade复合体的atp酶缺陷型突变体来通过成对的切口诱导靶向基因组缺失
[0986]
本实施例示出了cas3 atp酶缺陷型突变体蛋白(mcas3蛋白)如何能够用于在基因组dna的相对链上促进成对的切口以诱导靶向缺失。
[0987]
a.mcas3蛋白/ecocascade和mcas3蛋白-ecocascade rnp复合体dna模板组分的产生
[0988]
使得最小crispr阵列将两种大肠杆菌cascade(ecocascade)(seq id no:1871)rnp复合体靶向人类基因组中的gdna的相对链上的临近的位点。设计了大肠杆菌d452a mcas3蛋白(mcas3[d452a]),一种无解旋酶活性并因此仅具有切口活性的atp酶缺陷型变体(参见,例如mulepati,s.,et al.,j.biol.chem.288:22184
–
22192(2013)),以在ecocascade rnp复合体募集后经由成对的切口诱导靶向缺失。mcas3[d452a]蛋白被表达为与ecocascade(seq id no:1900)分离的单一组分,或表达为通过17个氨基酸的多肽连接子连接至ecocascade rnp复合体(seq id no:1901)内的cas8蛋白的融合蛋白。当将mcas3[d452a]蛋白表达为单一组分时,编码序列提供在表达载体上,其中其表达处于cmv启动子的控制下。cas3[d452a]蛋白/ecocascade是指表达为与ecocascade分离的组分的mcas3[d452a]蛋白。mcas3[d452a]蛋白
–
cascade rnp是指作为连接至ecocascade rnp复合体内的cas8蛋白的融合蛋白的mcas3[d452a]。将mcas3[d452a]蛋白
–
cascade rnp蛋白组分编码基因克隆至包含以下成分的载体中:使得能够在哺乳动物细胞中进行递送和表达的cmv启动子、经由2a“核糖体跳跃”序列连接的cas基因,以及利用17-aa连接子(seq id no:1901)附接至cas8以制备mcas3[d452a]-cas8融合蛋白的cas3的atp酶缺陷型突变体变体(d452a)。当将mcas3[d452a]蛋白表达为与cas8蛋白的融合蛋白时,融合蛋白组装为ecocascade rnp复合体(mcas3[d452a]蛋白
–
ecocascade rnp复合体)的一部分。
[0989]
两个向导靶序列(向导偏移量)之间的距离在1bp和120bp之间。确定了ecocascade rnp复合体的方向,使得pams相对于向导rna靶序列朝向内(pam在内)或朝向外(pam在外)。与核酸靶序列相关的pam序列选自以下:aat、ata、aac、aaa、gag、atg、agg或aag。
[0990]
使用3种寡核苷酸(seq id no:1513to seq id no:1515)和编码使得cascade rnp能够靶向临近的位点的“重复区-间隔区-重复区-间隔区-重复区”序列的独特的引物,利用
基于pcr的组装产生了最小crispr阵列。所得的扩增子将包含驱动包含向导的编码序列的最小crispr阵列的表达的hu6启动子(参见,例如实施例20a;图42a)。基本上按照制造商的说明,使用(beckman coulter,pasadena,ca)珠子纯化和浓缩pcr组装的最小crispr阵列。
[0991]
b.编码foki-cascade rnp复合体组分的载体的转染
[0992]
基本上按照实施例8b中所述进行转染条件,具有以下修改。进行核转染之前,将5μl含有dna模板的溶液转移至96孔板的单个孔中。对于mcas3[d452a]蛋白/ecocascade rnp复合体表达,孔包含1.5μg的编码mcas3[d452a]蛋白的质粒和1.5μg的编码ecocascade的质粒,以及0.3μg的编码最小crispr阵列的线性pcr产物。对于mcas3[d452a]蛋白
–
ecocascade rnp复合体表达,孔包含3μg的编码mcas3[d452a]
–
ecocascade蛋白(包括mcas3[d452a]-cas8融合蛋白)的质粒,以及0.3μg的编码最小crispr阵列的线性pcr产物。
[0993]
c. 来自转染的细胞gdna的深度测序
[0994]
测试了包含多个靶标位点的六个基因座在gdna的相对链上的成对的缺口(hzgj基因座,30个靶标位点;nphp3-acad11基因座,60个靶标位点;jak1基因座1,49个靶标位点;jak1基因座2,33个靶标位点;nmnat2基因座,38个靶标位点;和erbb2基因座,26个靶标位点)。针对包含指导cascade复合体结合至靶标位点的向导的mcas3[d452a]蛋白/ecocascade rnp复合体和mcas3[d452a]蛋白
–
ecocascade rnp复合体,测试了gdna的相对链上的成对的切口。
[0995]
基本上按实施例8c所述进行深度测序,并按实施例8d所述进行分析,所不同的是对上述靶标使用了对应于上述靶标的不同的靶标特异性引物。
[0996]
表50显示了靶向所示的靶标位点的mcas3[d452a]蛋白
–
ecocascade rnp复合体的30个hzgj靶标位点上的示例性的编辑数据。图48显示了利用mcas3[d452a]/ecocascade或mcas3[d452a]
–
ecocascade的在30个hzgj靶标位点处的示例性的基因组编辑数据。在图48中,垂直轴是%插入缺失,且水平轴是bp的间隔区间距。这里,对于每对cascade复合体,一种rnp固定在特定的靶标位点处,且第二种rnp在一系列距离内于不同的靶标位点处导向上游或下游。在图48中,连接它们的黑色圆圈和黑色线对应于利用mcas3-ecocascade的编辑,并且连接它们的灰色圆圈和灰色线对应于利用mcas3/ecocascade的编辑。对于绝大多数位点,利用mcas3/ecocascade的编辑低于检测限,而利用mcas3-ecocascade的编辑的范围从低于检测限至高达~4%插入缺失。mcas3-ecocascade使得能够在一系列向导rna偏移范围内进行靶向缺失,但利用pam在外配置最高。
[0997]
表50成对切口编辑数据
[0998]
[0999][1000]
基本上按照与本实施例中所示相同的方案,来自另外的位点的数据表明,当cascade rnp复合体以pam在外配置定向时,利用mcas3[d452a]
–
ecocascade样品可获得最佳的基因组编辑。利用mcas3[d452a]/ecocascade,高于检测限的编辑在26/238个靶标位点处可见,并且超过0.1%的编辑在1/238个靶标位点中可见(即对于大多数位点,低于检测限),而利用mcas3[d452a]
–
ecocascade,高于检测限的编辑在128/242个靶标位点处可见,并且高于0.1%的编辑在1/238个靶标位点处可见。mcas3[d452a]
–
ecocascade使得能够在一系列向导偏移量内进行靶向缺失,其中当cascade rnp复合体为pam在外配置时最高。
[1001]
本实施例中的数据显示包含mcas3蛋白的cascade rnp复合体可以用于在gdna的相对链上提供成对的切口,并因此促进宿主细胞(例如,人类细胞)的基因组中的靶向缺失。
[1002]
实施例26
[1003]
用于产生基因组缺失的cas3 atp酶缺陷型突变体
[1004]
本申请中描述了几种限制和/或限定由与cas3蛋白相关的cascade rnp复合体所促进的缺失长度的方法。本实施例示出了不成对的atp酶缺陷型突变体cas3蛋白如何能够
用于产生靶向基因组缺失;因此,使用单一cascade rnp复合体在单个位点处提供切口。
[1005]
a.用于转染至靶细胞中的假单胞菌s-6-2cas3变体和psecascadernp复合体组分的产生
[1006]
最小crispr阵列被设计为将假单胞菌s-6-2cascade(psecascade)rnp复合体靶向人类基因组中trac位点中的8个靶标(seq id no:1902至seq id no:1909)。这些序列示出在表51中。
[1007][1008]
基本上按实施例25a中所述,使用3种寡核苷酸(seq id no:1513至seq id no:1515)和编码使得能够进行psecascade rnp复合体靶向的“重复区-间隔区-重复区-间隔区-重复区”序列的独特的引物,利用基于pcr的组装产生了最小crispr阵列。对于该最小crispr阵列,两种间隔区序列是相同的。用于产生最小crispr阵列的全套的寡核苷酸序列示出在表52中。
[1009][1010]
主要根据制造商的说明,使用(beckman coulter,pasadena,ca)珠子纯化和浓缩pcr组装的向导。
[1011]
设计了无atp酶/解旋酶活性并因此仅具有切口活性(称为mpsecas3;seq id no:1919)的假单胞菌s-6-2cas3(psecas3;seq id no:1918)的d448aatp酶突变体变体,以诱导靶向缺失。作为参考点,还产生了psecas3(seq id no:1920)的d75a核酸酶失活变体(称为dpsecas3*),以及psecas3(seq id no:1921)的atp酶核酸酶双突变体变体(称为dblmpsecas3)。每种靶标的pam序列是aag。
[1012]
将psecascade rnp复合体蛋白组分编码基因以及突变体psecas3基因克隆至包含cmv启动子的载体中,以使得能够在哺乳动物细胞中进行递送和表达。经由2a“核糖体跳跃”序列连接psecascade rnp复合体cas基因,并且所述由基因均包含n端nls序列以将编码的蛋白导向核。序列示出在表53中。
[1013][1014]
b.编码foki-cascade rnp复合体组分的载体的转染
[1015]
基本上按实施例8b所述进行转染条件,具有以下修改。在进行核转染之前,将6μl的含有dna模板的溶液转移至96孔板的单个孔中,其中孔包含3μg的编码psecascade蛋白组分的质粒、0.2μg的编码最小crispr阵列的线性pcr产物,以及1μg的编码mpsecas3、dpsecas3*或dblmcas3的质粒。
[1016]
c.来自转染的细胞的gdna的深度测序
[1017]
基本上按实施例8c中所述进行深度测序。然而,代替来自实施例8c的表36的引物y和z,使用了trac1至trac8靶标位点中的每个的正向和反向靶标特异性引物,以及miseq试剂盒v3,600个循环(illumina,san diego,ca)。
[1018]
图49显示了利用与mpsecas3、dpsecas3*或dblmcas3(n=2)中的每个相关的psecascade rnp复合体在8个trac靶标位点处的基因组编辑。在图49中,垂直轴为%编辑,且水平轴表示trac位点中的靶标位点。条沿水平轴的顺序是mpsecas3(黑色条)、dpsecas3*(灰色条)和dblmcas3(条纹条)。利用dpsecas3*或dblmpsecas3 psecascade rnp复合体很少在靶标位点处观察到编辑,但利用mpsecas3 psecascade rnp复合体在靶标位点处达到高达~7%基因组编辑,如通过缺失所检测到的。这些数据表明,没有atp酶/解旋酶活性因此仅具有切口活性的mpsecas3蛋白可以与psecascade rnp复合体一起使用在单个靶标处(即,不以成对的切口配置),从而在预期的切割位点处产生缺失。
[1019]
如对本领域技术人员显而易见的是,在不脱离本发明的精神和范围的情况下,可以对上述实施方案进行各种修改和改变。这样的修改和改变在本发明的范围内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签:

 热门咨询
热门咨询




tips