
 商标分类
商标分类  商标转让
商标转让 
一种植物高效胞嘧啶单碱基编辑器及其构建与应用的制作方法
 2021-02-02 06:02:24|
2021-02-02 06:02:24| 411|
411| 起点商标网
起点商标网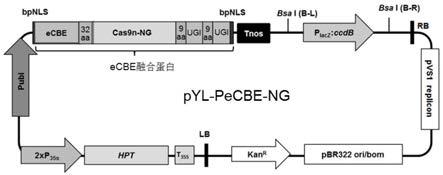
[0001]
本发明属于植物生物技术领域,具体涉及一种植物高效胞嘧啶单碱基编辑器及其构建与应用。
背景技术:
[0002]
作物的重要农艺性状往往由关键基因的点突变或单核苷酸多态性(snp)决定。虽然crispr/cas9介导的基因组编辑可以通过非同源末端修复(nhej)的方式在目标位点造成双链断裂(dsb)高效地敲除目标基因,但由于植物中同源直接修复(hdr)途径的低效性,并且dna供体模板传递到细胞也低效,因此想通过hdr的途径在特定位点精准插入和替换片段仍然有较大难度(chen et al.,2019,annual review of plant biology,70:667
–
697)。然而,基于crispr/cas系统开发的胞嘧啶碱基编辑器(cbes)和腺嘌呤碱基编辑器(abes)(komor et al.,2016,nature,533:420-424;rees and liu.,2018,nature reviews genetics,19:770-788)不需要供体模板及不造成双链dna断裂的条件下分别实现c-t(g-a)或a-g(t-c)的碱基替换。因此,单碱基编辑技术是植物基因功能研究和作物遗传改良的有效策略。
[0003]
目前,不同科研团队通过来自不同来源的胞嘧啶脱氨酶(如rapobec1、pmcda1和ha3a)与cas9缺刻酶变体(cas9n,含d10a氨基酸变异)融合,开发出多个不同的cbes系统。早期版本的cbe系统,例如:be1、be2、be3和be4在植物细胞中的编辑效率较低(平均大约0~30%)(ren et al.,2017,science china life sciences,60:516-519;hua et al.,2019,molecular plant,12:1003-1014;zeng et al.,2020,plant biotechnology journal,18:1348-1350)。新版本cbe如be4max使用apobec家族中祖先节点anc689胞嘧啶脱氨酶、bpnls和genscript公司优化的密码子,虽然在植物中的编辑效率得到进一步的提升(平均大约50%)(wang et al.,2019,plant biotechnology journal,17:1697-1699),但是由于rapobec1本身对靶点序列环境的偏好性,更易于编辑tc、cc位点(下划线为被编辑碱基),而几乎不编辑gc,因此编辑效果极不稳定。将激活诱导胞嘧啶脱氨酶(aid)家族的pmcda1脱氨酶与cas9n相融合,所得的target-aid编辑系统一定程度上比be3和be4高效(平均大约40%),并扩大了编辑窗口(c
2-c
12
)(wu et al.,2019,frontiers in genetics,10:379;zhong et al.,2019,molecular plant,12:1027-1036)。此外,通过将人的apobec3a脱氨酶(ha3a)和cas9n融合,所得的ha3a-pbe也一定程度提高了植物基因组的编辑效率(平均大约40%),特别是gc目标位置的替换效率(zong et al.,2018,nature biotechnology,36:950-953)。虽然在改良植物的碱基编辑系统中已经做了很大的努力,但目前cbe编辑系统在植物中的编辑仍存在活力较低,对靶点序列环境有较强的偏好性等问题。此外,由于大多数cbe系统都是融合识别ngg-pam的cas9n,降低了基因组靶向的选择性,这些因素大大限制了cbe在植物系统中的广泛应用。
[0004]
综上所述,开发适用于植物的高效、无序列偏好性以及广靶向的新的植物胞嘧啶单碱基编辑器是基因编辑技术优化的一个重要方向。
技术实现要素:
[0005]
植物胞嘧啶单碱基编辑效率低,常用cbes靶序列偏好性较强、靶点选择受限于ngg-pam,往往导致目标碱基c不能被成功编辑为t,从而限制了cbes在植物中的广泛应用。
[0006]
本发明的首要目的是提供一种植物高效胞嘧啶单碱基编辑器。
[0007]
本发明的首要目的是提供上述植物高效胞嘧啶单碱基编辑器的构建方法。
[0008]
本发明的首要目的是提供上述植物高效胞嘧啶单碱基编辑器的应用。
[0009]
本发明的目的通过以下技术方案实现:
[0010]
一种spcas9变体融合蛋白,命名为pecbe-ng,包含胞嘧啶脱氨酶ecbe、spcas9变体cas9n-ng和尿嘧啶糖基化酶抑制蛋白(ugi)。
[0011]
所述胞嘧啶脱氨酶ecbe的氨基酸序列如seq id no.1所示;所述spcas9变体cas9n-ng的氨基酸序列如seq id no.2所示;所述尿嘧啶糖基化酶抑制蛋白的氨基酸序列如seq id no.3所示。
[0012]
优选的,所述spcas9变体融合蛋白还包含以下序列中的一种或多种:接头(linker),核定位信号(nls),以及为了构建融合蛋白、促进重组蛋白的表达、获得自动分泌到宿主细胞核的重组蛋白、或利于重组蛋白的纯化而引入的氨基酸残基或氨基酸序列。
[0013]
所述接头可包括不干扰融合蛋白的功能的任何序列,优选为柔性接头。
[0014]
所述核定位信号的氨基酸序列如seq id no.4所示。
[0015]
所述核定位信号的个数优选为两个,且分别融合在所述spcas9变体融合蛋白的n端和c端。
[0016]
优选的,所述尿嘧啶糖基化酶抑制蛋白的个数为两个,且两个尿嘧啶糖基化酶抑制蛋白通过接头串联。
[0017]
更优选的,所述spcas9变体融合蛋白,从n端到c端,依次含1个核定位信号,1个胞嘧啶脱氨酶ecbe,1个spcas9变体cas9n-ng,2个串联的尿嘧啶糖基化酶抑制蛋白,以及1个核定位信号。
[0018]
最优选的,所述spcas9变体融合蛋白的氨基酸全序列如seq id no.5所示。其可直接通过化学合成方法得到,也可通过基因工程方法表达得到。
[0019]
一种多核苷酸序列,为编码上述spcas9变体融合蛋白的多核苷酸序列。
[0020]
优选的,所述多核苷酸序列如seq id no.6所示。所述序列是按照水稻的密码子进行优化得到。
[0021]
一种植物高效胞嘧啶单碱基编辑器,含有编码所述spcas9变体融合蛋白的多核苷酸序列,是将编码所述spcas9变体融合蛋白的多核苷酸序列整合至植物转化载体上得到。
[0022]
优选的,所述植物转化载体为双元表达载体,包括但不限于pcambia1300和在其基础上进行改造得到的载体,比如pylcrispr/cas9pubi-h,所述多核苷酸序列可插入至载体的pst i和bamh i酶切位点之间。
[0023]
上述植物高效胞嘧啶单碱基编辑器的构建方法,包括如下步骤:
[0024]
s1、分别合成编码nls-ecbe-linker 1的基因片段1,编码linker 2-ugi-linker 3-ugi-nls的基因片段2,并通过pcr反应在基因片段1的5
’
端添加一酶切位点,在基因片段2的3
’
添加另一酶切位点;克隆得到编码cas9n-ng的基因片段3;
[0025]
s2、通过重叠pcr技术,将带酶切位点的基因片段1的c端与基因片段3的n端连接,
得到融合蛋白nls-ecbe-linker 1-cas9n-ng的基因片段4;再将基因片段4的c端与将带酶切位点的基因片段2的n端连接,得到融合蛋白nls-ecbe-linker1-cas9n-ng-linker2-ugi-linker3-ugi-linker-nls的基因片段5;
[0026]
s3、将基因片段5插入至载体pylcrispr/cas9pubi-h两个相应的酶切位点之间,转化宿主菌,提取阳性质粒,测序,获得稳定的植物高效胞嘧啶单碱基编辑系统。
[0027]
优选的,步骤s3中将基因片段5插入载体通过gibson组装技术实现。
[0028]
优选的,步骤s3中所述的宿主菌为大肠杆菌top10f
’
。
[0029]
上述spcas9变体融合蛋白、或上述多核苷酸序列、或上述植物高效胞嘧啶单碱基编辑器在植物基因工程中的应用。尤指是在植物基因组单碱基编辑中的应用。所述植物包括但不限于单子叶植物,尤其是水稻。
[0030]
优选的,所述的应用具体操作如下:
[0031]
(1)确定待编辑基因的靶位点,根据靶位点设计并合成sgrna表达盒元件;
[0032]
(2)将所述sgrna表达盒元件整合至所述植物高效胞嘧啶单碱基编辑器上,得到靶基因胞嘧啶碱基替换载体;
[0033]
(3)将所述靶基因胞嘧啶碱基替换载体转化宿主细胞,筛选,获得相应的胞嘧啶碱基替换细胞。
[0034]
本发明适用于植物的高效、无序列偏好性、编辑窗口适合、脱靶效率低和广靶向的胞嘧啶碱基编辑器pyl-pecbe-ng,其spcas9变体融合蛋白pecbe-ng的表达盒从5
’
至3
’
端,依次是植物高表达组成型启动子ubi,spcas9变体融合蛋白pecbe-ng(核定位信号bpnls,ecbe胞嘧啶脱氨酶,柔性连接序列linker1(编码32aa),cas9n-ng突变体,柔性连接序列linker 2(编码9aa),尿嘧啶糖基化酶抑制蛋白ugi,柔性连接序列linker 3(编码9aa),尿嘧啶糖基化酶抑制蛋白ugi,核定位信号bpnls)和终止子tnos序列。编码spcas9变体融合蛋白pecbe-ng的碱基序列,全部由金斯瑞(中国武汉)公司按水稻密码子偏好性优化合成。
[0035]
本发明上述构建的pyl-pecbe-ng胞嘧啶单碱基编辑系统具有高的c-t的替换效率,且无靶点序列偏好性,主要编辑活性窗口在c
3-c8之间,自靶向t-dna的编辑效率低,脱靶效率也较低。克服了使用脱氨酶rapobec1的传统的be3和be4编辑器编辑效率低、强靶点偏好性等缺点,更有利于植物基因功能的研究和作物的遗传改良。
[0036]
含有所述的spcas9变体融合蛋白pecbe-ng的胞嘧啶单碱基编辑系统的质粒载体等材料,以及所述的pecbe-ng胞嘧啶单碱基编辑器在基因工程中的应用,均应在本发明的保护范围之内。
[0037]
更具体地,所述的pyl-pecbe-ng胞嘧啶单碱基编辑器系统能实现高效的c-t的单碱基替换,尤其是适用于基因功能筛选、大规模饱和突变、编辑调控元件、引入提前终止密码子或进行可变剪接等基因功能研究,与农作物遗传改良。
[0038]
本发明具有以下有益的效果:
[0039]
本发明提供了一种高效、无靶点序列偏好性、编辑窗口广、自靶向自身sgrna效率低、非c-t副产物少、脱靶效率低、靶向范围广、且可以多靶点编辑的植物cbe单碱基编辑系统。相比于其他目前已有的编辑系统,pyl-pecbe-ng的编辑更有优势。
附图说明
[0040]
图1为本发明植物胞嘧啶单碱基编辑器pyl-pecbe-ng载体结构示意图。
[0041]
图2为pyl-pecbe-ng和be4编辑器pyl-rac1-ng编辑效率对比结果图;其中,a为具有不同靶位点的pyl-pecbe-ng与pyl-rac1-ng的图谱,b为pyl-pecbe-ng和pyl-rac1-ng在9个靶点中的编辑效率对比结果(括号内为突变的植株数比总植株数,靶点中的pam用下划线和加粗凸显)。
[0042]
图3为pyl-pecbe-ng和pyl-rac1-ng的靶点偏好性和突变类型对比分析结果图;其中,a为从测试的9个靶点中统计ngg和ng靶点的平均编辑效率结果,b为从稳定编辑的c
3-c8窗口区间发生所有突变中分析gc,ac,tc,cc(c为被编辑的碱基)的偏好性结果,c为突变类型分析结果(ho表示homozygous mutation(纯合突变);he表示heterozygous mutation(杂合突变);trans表示transversions mutation(颠换),indels表示insertions or deletions(插入或删除)。
[0043]
图4为pyl-pecbe-ng和pyl-rac1-ng活性窗口比较结果图。
[0044]
图5为pyl-pecbe-ng自靶向编辑效率分析结果图。
[0045]
图6为pyl-pecbe-ng脱靶效率分析结果图(on-target靶序列差异的碱基用下划线和加粗凸显,靶点的pam加粗凸显)。
具体实施方式
[0046]
以下结合具体实施例和附图来进一步说明本发明,但实施例并不对本发明做任何形式的限定。
[0047]
除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。下述实施例中所使用的试验方法如无特殊说明,均为常规分子生物学方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
[0048]
实施例1植物胞嘧啶单碱基编辑器pyl-pecbe-ng的构建
[0049]
脱氨酶ecbe、2
×
ugi和bpnls的蛋白序列根据thuronyi等公布的序列(thuronyi et al.,2019,nature biotechnology,37:1070-1079),按bpnls-ecbe-linker 1和linker 2-ugi-linker 3-ugi-bpnls两个片段组合,由武汉genecreate公司按照水稻密码子偏好优化核酸序列直接合成(bpnls-ecbe-linker 1的核苷酸如seq id no.6的第4~636位碱基所示,linker 2-ugi-linker 3-ugi-bpnls的核苷酸如seq id no.6的第4738~5361位碱基所示)。直接使用申请人已有的优化合成的cas9n-ng变体(zeng et al.,2020,plant biotechnology journal,18:1348-1350)(如seq id no.6的第637~4737位碱基所示)用于融合。将优化合成的bpnls-ecbe-linker 1和linker 2-ugi-linker 3-ugi-bpnls通过overlaping pcr分别连接到cas9n-ng的两侧,形成完整融合的pecbe-ng,再用gibson组装的方法将其克隆到双元载体pylcrispr/cas9pubi-h(ma et al.,2015,molecular plant,8:1274-1284)的pst i和bamh i之间,最终获得高效、广靶向编辑器,命名为pyl-pecbe-ng。以申请人以前开发的be4编辑器pyl-cas9n-ng-cbe(简称pyl-rac1-ng)(zeng et al.,2020,plant biotechnology journal,18:1348-1350)作为编辑效率比较的对照。
[0050]
表1用于pyl-pecbe-ng的基础载体改造用引物
[0051]
引物名引物序列(5
’
—3
’
)
linker1-cas9n-ng-linker2-ugi-linker3-ugi-linker-bpnls融合片段(简称ecbe-cas9n-ng-2
×
ugi融合片段)。
[0066]
pcr体系(50μl):2
×
phanta max buffer 25μl,10mmol/l dntps mix 1.0μl,phanta max polymerase 1.0μl,第一轮扩增的linker 2-ugi-linker 3-ugi-bpnls片段n和第二轮扩增的ecbe-cas9n-ng融合dna片段各取1.0μl,10μmol/l f-pecbe-ng-1 1.0μl,10μmol/l r-pecbe-ng-4 1.0μl,ddh2o补足到50μl。
[0067]
pcr程序:预变性95℃2min,28个pcr循环(95℃10s,56℃15s,72℃4.5min),延伸72℃2min。
[0068]
用genstar纯化试剂盒,纯化扩增的bpnls-ecbe-linker1-cas9n-ng-linker2-ugi-linker3-ugi-bpnls融合dna片段的pcr产物。用pst i和bamh i酶切pylcrispr/cas9pubi-h(ma et al.,2015,molecular plant,8:1274-1284):10
×
faster digest buffer,pst i 0.5μl,bamh i 0.5μl,pylcrispr/cas9pubi-h 300ng,ddh2o补足到10μl,37℃反应1h,胶回收载体骨架,用于gibson组装反应(neb#e5510s):2
×
mix 5μl,ecbe-cas9n-ng-2
×
ugi融合片段60ng,胶回收载体骨架90ng,ddh2o补足到10μl,50℃反应50min。取1.5μl gibson的连接产物,电激转化大肠杆菌top10f
’
,在卡那霉素抗性(kana)lb平板上,筛选转化单克隆。并将阳性克隆送测序,从而获得pyl-pecbe-ng基础载体质粒。
[0069]
实施例2 pyl-pecbe-ng具有更高的胞嘧啶碱基编辑效率
[0070]
参考本发明人团队前期发表的文献(ma et al.,2015,molecular plant,8:1274-1284;ma and liu,2016,current protocols in molecular biology,115:31.6.1-31.6.21;曾栋昌等,2018,中国科学:生命科学,48:783-794),分别构建小核rna基因启动子(osu6a、osu6b、osu6c和osu3)驱动不同靶点的sgrna表达盒,用“金门组装,golden gate”方式插入pyl-pecbe-ng双元载体(ma et al.,2015,molecular plant,8:1274-1284),转化水稻,并对转化体靶点测序,分析pyl-pecbe-ng编辑效率。具体操作如下:
[0071]
1.t1~t9的靶点引物设计
[0072]
利用我们开发的网上程序crispr-ge网页(http://skl.scau.edu.cn/)(xie et al.,2018,molecular plant,11:720-735),进入引物设计(primerdesign)子程序,primerdesign-v分支程序,选择对应启动子,勾上复选框,并选择method2,点击design,自动生成9个靶点引物gr-t#与u#-t#(表2)。
[0073]
2.t1~t9的sgrna表达盒的overlapping pcr拼接
[0074]
按照我们前期发表的文献(ma et al.,2015,molecular plant,8:1274-1284;ma and liu,2016,current protocols in molecular biology,115:31.6.1-31.6.21;曾栋昌等,2018,中国科学:生命科学,48:783-794)操作,通过两轮pcr,获得两侧具有bsa i酶切位点的小rna启动子驱动的sgrna表达盒。
[0075]
表2第一轮pcr靶点引物gr-t#与u#-t#序列
[0076]
引物名序列(5
’---3’
)grt1acccccccacaggctcgcgagttttagagctagaaat(seq id no.13)osu6at1tcgcgagcctgtgggggggtcggcagccaagccagca(seq id no.14)grt2tcgtcggcggcgatggtgagttttagagctagaaat(seq id no.15)osu6at2tcaccatcgccgccgacgacggcagccaagccagca(seq id no.16)
grt3accgccaccgtcgtcgccaagttttagagctagaaat(seq id no.17)osu6bt3ttggcgacgacggtggcggtcaacacaagcggcagc(seq id no.18)grt4tgcatgcatgcacccatgcgttttagagctagaaat(seq id no.19)osu3t4gcatgggtgcatgcatgcatgccacggatcatctgc(seq id no.20)grt5atctctgcactgaattgaatgttttagagctagaaat(seq id no.21)osu6at5attcaattcagtgcagagatcggcagccaagccagca(seq id no.22)grt6tccaccatgcaccacgacgtgttttagagctagaaat(seq id no.23)osu6at6acgtcgtggtgcatggtggacggcagccaagccagca(seq id no.24)grt7agctcaagctccgcgccgcgttttagagctagaaat(seq id no.25)osu6bt7gcggcgcggagcttgagctcaacacaagcggcagc(seq id no.26)grt8atcagcgaccggatctccccgttttagagctagaaat(seq id no.27)osu6bt8ggggagatccggtcgctgatcaacacaagcggcagc(seq id no.28)grt9cattctcccagttcttcgcgttttagagctagaaat(seq id no.29)osu3t9gcgaagaactgggagaatgtgccacggatcatctgc(seq id no.30)u-fctccgttttacctgtggaatcg(seq id no.31)gr-rcggaggaaaattccatccac(seq id no.32)
[0077]
第一轮pcr,利用设计的u#-t#/gr-t#引物(seq id no.13~seq id no.30,表2)将靶点序列引入到osu6/osu3启动子下游和sgrna序列的上游。在一个pcr体系中,利用u-f引物(seq id no.31,表2)与gr-t#引物配对,pcr扩增获得含靶点的启动子序列;利用gr-r引物(seq id no.32,表2)与u#-t#引物配对,pcr扩增获得含靶点的sgrna序列。pcr体系(20μl):2
×
phanta max buffer 10.0μl,10mmol/l dntps mix 0.4μl,phanta max polymerase 0.3μl,pylgrna-osu6/3(含启动子和sgrna质粒)(ma et al.,2015,molecular plant,8:1274-1284)3ng,10μmol/l u-f 0.4μl,10μmol/l gr-t#0.2μl,10μmol/l grna-r0.4μl,10μmol/l u#-t#0.2μl,ddh2o补足到20μl。pcr程序:预变性95℃1min,28个pcr循环(95℃10s,58℃15s,72℃20s),延伸72℃1min。
[0078]
表3构建多个sgrna表达盒的通用引物
[0079][0080]
注1:bsa i酶切末端设计成非回文序列,可产生高效的连接(golden gate ligation)。
[0081]
注2:如果连接多于8个sgrna表达盒,需要自行设计更多组pgs和pps引物,每组含有互补的非回文bsa i酶切末端。
[0082]
表4组装不同数量sgrna表达盒的第二轮pcr引物组合
[0083][0084]
第二轮pcr,使用第二轮pcr引物pps和pgs(seq id no.33~seq id no.48,表3和表4)拼接小rna启动子驱动的sgrna表达盒,并在pcr产物两侧加上bsa i酶切位点。t1~t4作为一组,t5~t9作为一组,分别构建1个4靶点和5靶点的sgrna表达盒载体。pcr体系(50μl):2
×
phanta max buffer25.0μl,10mmol/l dntps mix 1.0μl,phanta max polymerase 1.0μl,10
×
稀释上一轮pcr产物1.0μl,4靶点分别使用的引物,10μmol/l的pps-l/pgs-2(t1),pps-2/pgs-3(t2),pps-3/pgs-4(t3),pps-4/pgs-r(t4)各1.0μl;5靶点分别使用的引物,10μmol/l pps-l/pgs-2(t1),pps-2/pgs-3(t2),pps-3/pgs-4(t3),pps-4/pgs-5(t4),pps-5/pgs-r(t5)各1.0μl,ddh2o补足到50μl,pcr程序同上述第一轮pcr。使用genstar纯化试剂盒纯化第二轮pcr产物。
[0085]
3.含不同靶点的sgrna表达盒敲除载体的构建
[0086]
使用基于bsa i酶切和连接的“金门”克隆方法,以“边切边连接”方式(ma and liu,2016,current protocols in molecular biology,115:31.6.1-31.6.21;曾栋昌等,2018,中国科学:生命科学,48:783-794),分别组装t1~t4,t5~t9;两组小核rna启动子驱动的sgrna表达盒分别克隆到双元载体pyl-pecbe-ng上(图2a)。15μl反应体系:10
×
cutsmart buffer 1.5μl,10mmol/l atp 1.5μl,pyl-pecbe-ng质粒80~100ng,纯化后的sgrna表达盒10~15ng,bsa i-hf 10u,t4 dna ligase 35u,ddh2o补足到15μl。用pcr仪变温循环进行酶切连接反应:37℃10min,接着10-12循环(37℃5min,10℃3min,20℃5min);最后37℃3min。透析连接产物后,电激转化进入dh10b细胞,在卡拉抗性(kan)lb板上筛选,用引物对sp-l1/sp-r(seq id no.49和seq id no.50,表3),根据文献(ma and liu,2016,current protocols in molecular biology,115:31.6.1-31.6.21;曾栋昌等,2018,中国科学:生命科学,48:783-794),做菌落pcr,筛选阳性克隆,最终用引物sp-l1(seq id no.49)测序确定。
[0087]
4.pyl-pecbe-ng具有较高的编辑效率。
[0088]
利用农杆菌介导的水稻(粳稻中花11)愈伤转化,把上述含有t1-t4和t5-t9两组不同小rna启动子驱动的sgrna的4靶点和5靶点载体转化水稻愈伤(图2a),提取t0代转化植株的叶片dna作为模板,使用amp和seq(seq id no.51~seq id no.77,表5)扩增单碱基编辑靶位点的dna片段,直接sanger测序,通过将测序结果和参考序列比对,统计、比较pyl-rac1-ng和pyl-pecbe-ng的编辑效率。结果表明pyl-pecbe-ng的平均编辑效率最高(62.6%),其中在t7靶点的编辑效率高达86.3%(图2b)。
[0089]
5.pyl-pecbe-ng在ng靶点中同样具有较高编辑效率,且无靶点序列偏好性,同时副产物较少。
[0090]
cas9-ng虽然扩大了基因组的编辑范围,但是和传统的spcas9相比,其切割编辑效率发生了一定幅度下降(zeng et al.,2020,plant biotechnology journal,18:1348-1350),为了探究pyl-pecbe-ng单碱基编辑器是否在ng靶点可以实现高效编辑,我们从9个靶点中统计了pyl-pecbe-ng在ngg靶点和ng靶点的平均编辑效率,结果显示,pyl-pecbe-ng在ngg的平均编辑效率虽然稍低于ng靶位点,但是在ng靶位点同样保持着较高的编辑效率(图3a)。根据t0代的测序结果,我们统计了所有测试的载体在c
3-c8活性窗口上gc、ac、tc和cc基序的平均编辑效率,并分析了它们的碱基偏好。统计结果显示,以be4为载体结构的rac1脱氨酶rac1-ng出现严重序列环境的偏好性,在tc(8.8%)和cc(5.2%)具有低编辑效率,而对gc和ac位点在9个靶点中都未发生编辑(图3b)。而pyl-pecbe-ng消除了靶点序列环境的偏好性,且具有较高的编辑效率。此外,pyl-pecbe-ng的编辑产物主要以纯合和杂合突变为主,而非c-t编辑的副产物只有2.9%(图3c)。
[0091]
表5 t0代转化植株靶点扩增及测序引物
[0092][0093]
6.pyl-pecbe-ng有适中的编辑窗口
[0094]
根据t0代的测序结果,我们统计了t1~t9中9个靶点在c-2
~c
15
每个位点中c-t的平均编辑效率,绘制成编辑活性窗口图。结果显示,pyl-pecbe-ng有比较适中的编辑窗口(c
3-c8)(图4)。适中编辑窗口的脱氨酶搭配广靶向的cas蛋白更有助于单碱基编辑技术在动植物基因组编辑中的运用。
[0095]
7.pyl-pecbe-ng自靶向sgrna靶点的效率低
[0096]
在以往的报道中,cas9-ng除了可以识别靶向ngg-pam外,还可以识别gtt-pam,因此cas9-ng除了靶向基因组靶点,同时还可能靶向t-dna自身的sgrna的靶点(qin et al.,2020,nat plants 6:197-201)。为了探究本研究的pyl-pecbe-ng自靶向t-dna自身sgrna靶位点的效率,直接扩增t0代转化苗的sgrna靶点送sanger测序,结果显示pecbe-ng只在t6靶点展现微弱的sgrna自编辑效率(6.9%),而在其他靶点均未检测到编辑(图5),9个靶点的平均自编辑效率远低于已知spcas9-ng大于50%的自靶向编辑效率。
[0097]
8.pyl-pecbe-ng的脱靶效率也较低
[0098]
利用我们课题组开发的网上程序crispr-ge网页(http://skl.scau.edu.cn/)(xie et al.,2018,molecular plant,11:720-735),进入引物设计(off-target)子程序,输入t2,t3,t4的靶点序列,从这三个靶点中选择具有≤3个碱基不匹配的脱靶位点用于脱靶效率分析。用抗性愈伤基因组dna为模板pcr扩增潜在的脱靶位点做高通量测序分析。结果显示,pecbe-ng除了在1个碱基不匹配的潜在脱靶位点检测到有脱靶的发生外,在2和3个
碱基不匹配的候选脱靶位点未发现脱靶(图6)。说明了pyl-pecbe-ng的由sgrna引起的脱靶效率低。
[0099]
综上所述,本发明开发的新型高效植物胞嘧啶碱基编辑器pyl-pecbe-ng比以前的cbe编辑系统(如申请人以前的be4编辑器pyl-rac1-ng),具有更与脱靶效率低等优点,能够广泛的用于作物基因功能筛选、大规模饱和突变、编辑调控元件、引入提前终止密码子或进行可变剪接等操作。
[0100]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips