
 商标分类
商标分类  商标转让
商标转让 
以高收率制备MMA的方法与流程
 2021-02-02 05:02:47|
2021-02-02 05:02:47| 223|
223| 起点商标网
起点商标网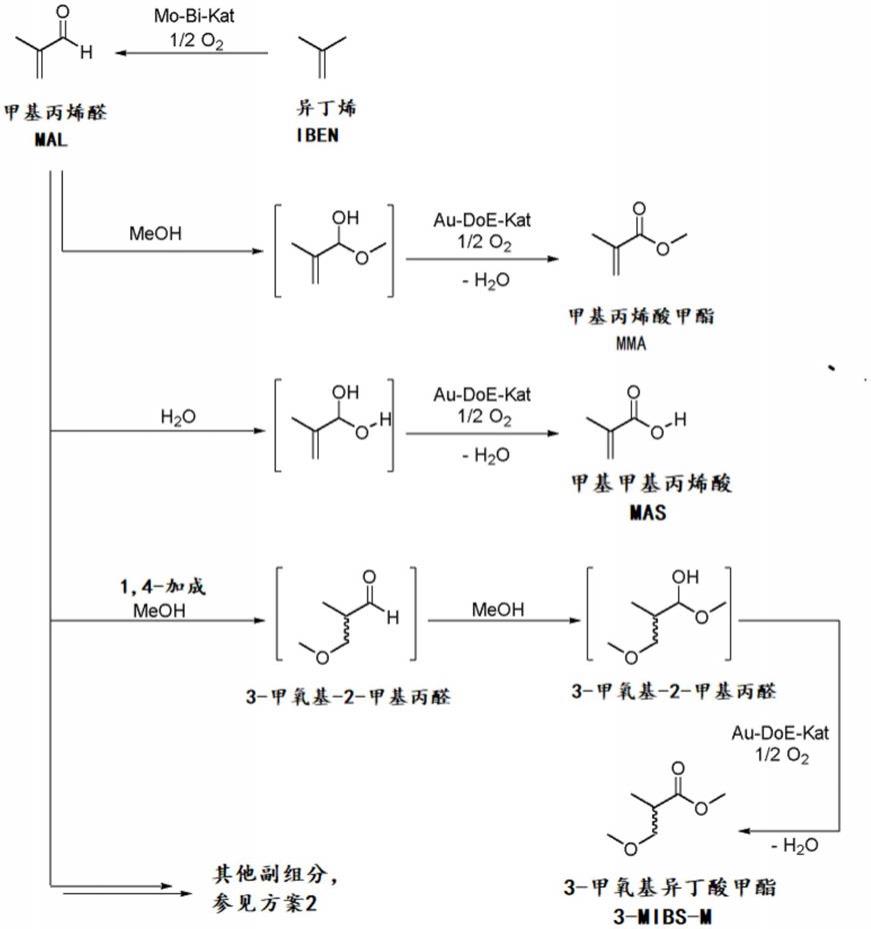
以高收率制备mma的方法
技术领域
[0001]
本发明涉及与现有技术相比具有提高的收率的通过甲基丙烯醛的直接氧化酯化制备甲基丙烯酸甲酯的方法。甲基丙烯酸甲酯大量用于制备聚合物和与其它可聚合化合物的共聚物。此外,甲基丙烯酸甲酯是可通过与适当的醇的酯交换制成的各种基于甲基丙烯酸(mas)的特种酯的重要合成单元。因此对用于制备这种原材料的尽可能简单、经济和环保的方法很感兴趣。
[0002]
更特别地,本发明涉及来自甲基丙烯醛的氧化酯化的反应器输出物的优化后处理,借此可分离出特定副产物,然后另外转化成甲基丙烯酸烷基酯,尤其是转化成mma。
背景技术:
[0003]
甲基丙烯酸甲酯(mma)当前通过由c2、c3或c4合成单元起始的各种方法生产。在这些方法的一种中,通过异丁烯或叔丁醇在非均相催化剂上用大气氧在气相中氧化以提供甲基丙烯醛(mal)和随后使用甲醇将甲基丙烯醛氧化酯化反应获得mma。asahi开发的这种方法尤其描述在公开文本us 5,969,178和us 7,012,039中。这种方法的缺点尤其是很高的能量需求。
[0004]
以下方案1示意显示了通过所谓的asahi工艺制备mma的过程,其由c4(异丁烯或叔丁醇)开始,中间分离出mal和随后用甲醇将mal氧化酯化(缩写为“doe”)以产生mma,作为副产物形成甲基丙烯酸(mas)。
[0005][0006]
方案1:由c4原材料制备mma的asahi工艺
[0007]
在该方法的发展中,在第一阶段中由丙醛和福尔马林获得甲基丙烯醛。这种方法描述在wo 2014/170223中。
[0008]
us 5,969,178描述了将异丁烯或叔丁醇氧化转化成甲基丙烯醛和随后氧化酯化成mma的方法。在这种第二阶段中,使具有降低的水含量的甲基丙烯醛和甲醇的液体混合物与分子氧和钯催化剂反应,其中所述催化剂多数在载体上的钯-铅催化剂形式。随后在第一蒸馏阶段中,在柱顶下方从氧化酯化的粗产物中分离出甲基丙烯醛和甲醇的混合物,同时从柱顶除去低沸点成分。然后将含mma的柱底物送入第二蒸馏阶段,其中从柱顶分离出甲醇和饱和烃的共沸物。将包含粗制mma的柱底物送往进一步后处理,同时借助相分离器和第三蒸馏柱从柱顶获得的馏分中分离甲醇并送回反应器。在此要注意的是,由于形成的共沸物,甲醇可含有相对大量的水并因此必须送往脱水过程。
[0009]
作为对这种方法的替代,us 5,969,178公开了在仅一个柱中的后处理,其中在所述柱中,进料必须位于柱底物上方。从该柱的柱顶除去反应器输出物中的低沸点成分。留在柱底物中的是粗制mma和水的混合物,将其送往进一步后处理。通过必须首先确定其确切位
置并可通过添加各种筛板调节所述位置的侧料流,最后从柱中取出甲基丙烯醛和甲醇的混合物,其意于送回反应器。us 5,969,178其中自己指出,由于各种共沸物,这种方法难以进行。此外,始终作为副产物存在的甲基丙烯酸在此特别起到重要作用。根据这种方法,尽管us 5,969,178没有提到这一问题,但甲基丙烯酸的分离除去方式使得其留在要送去处置的相中并且离析只有有限的吸引力。但是,这意味着这种方法的甲基丙烯酸系产物的总收率降低。
[0010]
us 7,012,039公开了略有不同的来自氧化酯化的反应器输出物的后处理。在此,在第一蒸馏阶段中,经由筛板从柱顶蒸馏出甲基丙烯醛并将来自柱底物的水性含mma混合物送入相分离器。在所述相分离器中,通过添加硫酸将该混合物调节到大约2的ph值。然后借助离心实现硫酸酸化的水与有机/油相的分离。这种有机相在进一步蒸馏中分离成高沸点成分和含mma的相,其从柱顶取出。然后在第三蒸馏中将含mma的相与低沸点成分分离。此后甚至接着第四蒸馏以最终提纯。
[0011]
这种方法的问题是硫酸,其需要大量添加并对装置部件可能具有腐蚀作用。因此,这些部件,特别例如相分离器或第二蒸馏柱,必须由对此合适的材料制成。此外,us 7,012,039也没有提及同时生成的甲基丙烯酸或留在产物中的残余甲醇的处理。但是可以推测,前者在蒸馏阶段中同时分离除去,而甲醇仅部分地可随甲基丙烯醛获得并送回,而剩余部分可能在第三蒸馏阶段中损失。
[0012]
wo 2014/170223描述了与us 7,012,039类似的方法。唯一区别在于,在实际反应中,在回路中通过添加氢氧化钠的甲醇溶液调节ph值。如果该方法没有在ph调节下进行,如在这两篇引用的公开文本中所述,该反应发生“酸化”。在低于7的ph范围内,这导致更多地形成甲基丙烯醛的缩醛,其必须以复杂的方式分离脱除或水解解离。此外,氧化催化剂的活性尤其取决于ph值等因素。在低于ph 7的条件下,随着反应基质(reaktionsmatrize)的ph值降低,催化剂表现出越来越低的活性,这是不理想的。ph调节的目的另外尤其在于保护催化剂。此外,由于盐内容物,在相分离中的水相脱除更简单。但是,这样的另一后果在于,形成的甲基丙烯酸为钠盐形式并稍后随水相脱除和弃置。在相分离中加入硫酸的变化方案中,回收游离酸,但代价在于产生硫酸(氢)钠,这在处置中可造成其它问题。
[0013]
原则上,并且在现有技术的总结中,在不饱和醛的直接氧化酯化中,甲基丙烯醛或还有丙烯醛在甲醇作为一般而言的醇存在下的氧化酯化产生各种高沸点组分。这些高沸物的沸点在比所需产物甲基丙烯酸甲酯(mma)高的水平,并且因此在mma的稍后离析中必须从mma中分离出以产生明显大于99重量%的合适的单体纯度。这些副产物,尤其是甲基丙烯酸、甲氧基异丁酸甲酯、二聚甲基丙烯醛(醛)和二聚甲基丙烯醛的相应的酯以显著的量形成并对所需mma的收率可能具有明显不利的影响。由于必须将它们从形成的mma中分离出,同样必须将这些物质送往处置操作,在最简单的情况下燃烧、热利用以回收蒸汽或在水处理厂中生物分解。
[0014]
副产物之一是甲基丙烯酸,其在水存在下的doe反应中形成,通常在该反应的连续运行过程中以2重量%至20重量%的稳态浓度存在于反应器中。如果doe反应在恒定ph下进行,则甲基丙烯酸在其形成时被碱或碱性辅助剂(在最简单的情况下为碱金属化合物)至少部分中和。
[0015]
该反应同样形成甲氧基异缩丁醛—甲醇与甲基丙烯醛的迈克尔加成产物。作为副
反应,这种甲氧基异缩丁醛在doe条件下在含氧气体和例如甲醇存在下反应,而至少部分产生甲氧基异丁酸甲酯(mibs-m)。该反应是碱催化的,即由于加入用于控制doe反应的ph的碱而不可避免地形成。一般观察到,随着ph提高,甲氧基异缩丁醛的形成和因此mibsm的后续形成变得更明显。根据所用催化剂和根据doe反应的所选稳态ph,在反应器中形成0.1%至5%的这种副产物。
[0016]
现有技术中,例如德国公开文本1279015中,等人描述了烷氧基丙酸烷基酯可如何热和催化解离,其中获得不饱和丙烯酸酯。在这种情况下,使用纯物质作为反应物并描述了纯β消除。
[0017]
在wo2016166525a1申请中,lucite描述了催化的、碱诱导的甲氧基异丁酸甲酯的解离成甲醇和甲基丙烯酸甲酯。在此,在保护气体气氛下在95℃下使用纯物质作为反应物。但是,转化率仅是很低的37%。
[0018]
以下方案2显示了反应矩阵(reaktionmatrix)(以乙烯和合成气和福尔马林生成甲基丙烯醛为例;如所述,也有可能从异丁烯或叔丁醇起始生成甲基丙烯醛);特别显示了目标产物mma和高沸点副产物mas、mibsm、dimal和dimal酯的形成:
[0019][0020]
方案2:用于制备mma的c2工艺的反应矩阵
[0021]
在doe反应的产物矩阵(produktmatrix)中获得包含水、甲醇、mma、游离甲基丙烯酸以及碱中和的甲基丙烯酸,例如甲基丙烯酸钠和方案2中所述的其它副产物的混合物。
[0022]
概括而言,现有技术方法的以下方面,尤其互相结合地,需要改进:
[0023]-mma的尽可能高的收率
[0024]-副产物甲基丙烯酸转化成甲基丙烯酸甲酯并离析
[0025]-副产物甲氧基异丁酸甲酯(mibsm)转化成甲醇和mma,并且形成的甲醇再循环,或mibsm用于交叉酯交换以将mas转化成mma
[0026]-二聚甲基丙烯醛和相应的二-mal酯至少部分转化成mma和甲基丙烯醛
[0027]-副产物的尽可能高程度再循环
[0028]-处置料流或废气的尽可能高清洁度
技术实现要素:
[0029]
技术问题
[0030]
考虑到现有技术,本发明要解决的技术问题因此是提供不受常规方法的缺点影响并带来比现有技术高的收率的用于甲基丙烯醛的氧化酯化的技术上改进的方法。
[0031]
本发明要解决的技术问题尤其是提供在来自用于获得mma的甲基丙烯醛和甲醇的氧化酯化的粗产物的后处理中的改进和因此与现有技术相比改进这种方法的总收率。
[0032]
此外,要解决的技术问题是在尽可能高的程度上分离出尽可能多的在该方法中形成的副产物,尤其是甲氧基异丁酸烷基酯和甲基丙烯酸,并以提高收率的方式将它们供应给甲基丙烯酸烷基酯的制备。本发明要解决的另一明确技术问题是找出有可能允许在单个工艺步骤中转化这些副产物中的多种以产生mma并优选在粗产物后处理中以以前对于mma分离需要的工艺步骤为基础的方法。更特别地,这涉及以由于其它原因已经存在的柱、相分离器、萃取器或通用设备为基础的对这些技术问题的解决方案。
[0033]
要解决的技术问题另外尤其是提供可用尽可能低的处置成本运行的方法,特别是通过减少废物料流中的有机成分和酸的生成。
[0034]
解决方案
[0035]
通过一种制备甲基丙烯酸烷基酯的方法实现了这些目的,其中在第一反应阶段中在反应器i中制备甲基丙烯醛,并且其在第二反应阶段中在反应器ii中用醇,优选用甲醇氧化酯化,以产生甲基丙烯酸烷基酯,优选相应地产生mma,其中所述方法根据本发明的特征在于a.将来自反应器ii的反应器输出物分离成含有主要部分的甲基丙烯酸烷基酯的馏分和含有甲基丙烯酸和烷氧基异丁酸烷基酯(aibsm)的第二馏分。在该醇是甲醇的优选情况下,aibsm是甲氧基异丁酸甲酯(mibs-m)。
[0036]
此外,根据本发明的方法的特征在于b.这种第二馏分在反应器iii中转化以由aibsm和甲基丙烯酸形成进一步的甲基丙烯酸烷基酯。该化学转化显示在方案3中,其中举例显示借助mas和mibs-m的反应:
[0037][0038]
方案3:a)由mal形成mibsm和mas的反应矩阵。b)交叉酯交换以获得mma。c)di-mal酯解离成mma和mal。
[0039]
非常有利地,在此可表明,副产物(mibs-m)的解离中释放甲醇,其是交叉酯交换以将第二副产物,即甲基丙烯酸mas转化形成mma所需的。痕量形成的3-甲氧基异丁酸也在这一反应中转化,首先转化成3-mibs-m,然后转化成mma。
[0040]
所有这些反应同步进行,优选在单个装置或单个反应器中,使得将许多副产物最终转化成目标产物mma。特别可在us 5,969,178、us 7,012,039和wo 2014/170223中查阅到包含上述两个反应阶段的用于合成mma的方法。根据本发明的可能的流程图在此显示在图1中。
[0041]
在此,用于合成甲基丙烯醛的该方法的第一阶段根据本发明可自由选择。根据本发明的方法不仅适用于基于叔丁醇或异丁烯的第一阶段合成,也适用于基于丙醛和福尔马
林的第一阶段合成。
[0042]
在基于丙醛和福尔马林的甲基丙烯醛制备中,原则上也有两种可能的工艺变化方案,它们根据本发明得到可用于doe反应的品质的甲基丙烯醛。首先,丙醛和福尔马林可在搅拌或循环泵送的反应器中在20℃至120℃的温度下在1巴至10巴的压力下转化。这需要大于10分钟的反应时间实现充分转化。其次,可由这些反应物制备mal,其中该反应在10至100巴的中等压力下在120℃至250℃的相对较高温度下以2秒至20秒的反应时间实现理想的高收率。
[0043]
优选在液相中在2至100巴的压力,优选2至50巴的压力和10℃至200℃的温度下用非均相催化剂进行氧化酯化。非均相催化剂通常包含具有小于20nm,优选0.2至20nm的粒子尺寸的负载型含金纳米粒子。反应阶段(a)可包含任选和较不优选的蒸馏柱ii以分离除去低沸物,如剩余丙醛,和/或高沸物,如二聚甲基丙烯醛。
[0044]
更优选地,在步骤a.中,借助至少一次萃取和/或蒸馏实施分离。在此也有可能使用多个蒸馏步骤或萃取步骤,也有可能使用至少一次蒸馏和至少一次萃取的组合。
[0045]
在根据本发明的方法中已经发现非常特别优选的是以在第一蒸馏柱中将来自反应器ii的反应器输出物脱除甲基丙烯醛和部分地脱除醇以在此获得包含甲基丙烯酸烷基酯、水、碱金属甲基丙烯酸盐和/或甲基丙烯酸、mibsm和醇的料流的方式进行该方法。随后,将这一料流与强酸混合并在萃取中分离成包含甲基丙烯酸烷基酯、mas和mibsm的疏水相,和包含水、醇和剩余相对少量的来自该反应的主要产物和副产物的亲水相。
[0046]
对此替代性地和同样优选地,以在第一蒸馏柱中将来自反应器ii的反应器输出物脱除甲基丙烯醛和部分地脱除醇以在此产生包含甲基丙烯酸烷基酯、水、碱金属甲基丙烯酸盐和/或甲基丙烯酸、mibsm和醇的料流的方式实施根据本发明的方法。在该方法的这一时刻,甲基丙烯酸可以是游离有机酸或碱金属盐的形式或游离酸和碱金属盐的混合物的形式。任选地,通过酸化,例如将该混合物与布朗斯台德酸混合,将盐形式的酸转化成游离酸。
[0047]
随后使这一料流经历萃取,以在此产生主要含有mma,还含有一定比例的有机副产物mas和mibsm的有机相。
[0048]
在萃取后获得的有机相在第二蒸馏中分离成包含甲基丙烯酸烷基酯和醇的低沸点相和包含水、mibsm和甲基丙烯酸的高沸点相。
[0049]
因此,在根据本发明的方法中,来自尤其在反应器ii中的doe反应的有价值产物mma与副产物的分离特别有利。更特别地,根据本发明因此令人惊讶地可实现mma与mas、mibsm、dimal和dimal酯和hibs异构体的分离。
[0050]
这种上述高沸点相随后,在本发明的第二个方面中,经历进一步反应,其中将doe的副产物转化成所需主要产物mma。在反应器iii中另外形成的mma显著提高该方法的总收率。有利地,将在反应器iii中获得的mma粗产物送往主工艺中的一个或多个后处理柱。
[0051]
反应器iii中的转化又是优选在至少90℃,更优选至少110℃,最优选120℃至170℃的温度下实施。因此,该反应可在不添加催化剂的情况下完全依靠热进行。更优选地,该反应在催化剂存在下依靠热进行,其中所述催化剂尤其可以是布朗斯台德酸。
[0052]
在第三种备选方案中,该转化也可在这样的催化剂存在下在低于130℃的温度下实施。
[0053]
更优选的是,布朗斯台德酸是强酸。根据本发明,强酸应被理解为是指比甲基丙烯
酸强的酸。这意味着这种酸具有在标准条件下比甲基丙烯酸小的pk
a
值。在这种情况下特别优选的无机酸是硫酸。较不优选的有机酸可以是例如甲磺酸或甲苯磺酸。另一合适的无机酸的实例是磷酸。硫酸在此已被发现是特别合适的催化剂。通常有利的是在反应器iii中使用与用于酸化和从碱金属甲基丙烯酸盐中释放mas的酸相同的催化剂酸。该酸在这种方法中的第三个功能是分解麻烦的甲基丙烯醛的缩醛。因此在最好的情况下能在该集成系统内只用单一一种酸就可以完成操作。这种普遍适用的酸的一个特别合适的实例是根据使用功能具有不同浓度或可调节到不同浓度的硫酸。
[0054]
进一步发现优选的是将来自反应器iii的反应器输出物与在分离脱除mibsm和甲基丙烯酸后获得的含甲基丙烯酸烷基酯的馏分合并供进一步后处理。此处的这种第二馏分一方面可以是在工艺步骤a)中的根据本发明的分离后的料流—并非含mibsm的料流。但是,更有利的是在合并这两个馏分以供进一步提纯之前首先在一个或多个步骤中提纯这一料流。预提纯的选择例如取决于为反应器i中的第一工艺步骤选择的工艺和其中所用的原材料。实际粗制甲基丙烯酸烷基酯的这种预提纯可以例如是高沸物柱、低沸物柱或串联的这两种蒸馏。对此替代性地或与其并行地,当然也有可能首先预提纯来自反应器iii的反应器输出物,然后再将这一馏分与上文提到的另一馏分合并。这种提纯也可能是一次或多次萃取或蒸馏或其组合。为此,获自该提纯的所用的酸可任选再循环回反应器iii。更优选地,在反应器ii的下游直接存在用于分离出mal的蒸馏柱。这随后可再循环到反应器ii或上游提纯步骤。
[0055]
在方法方面,构成本发明的一个方面的是在所述反应器i中制备甲基丙烯醛。ullmanns encyclopedia of industrial chemistry(乌尔曼工业化学大全),2012,wiley-vch verlag gmbh,weinheim,doi:10.1002/14356007.a01_149.pub2给出了甲基丙烯醛的制备方法和工艺的很好的综述。
[0056]
可以表明,多种方法变化方案原则上适用于制备甲基丙烯醛,它们可举例概括如下:
[0057]
a.由甲醛和福尔马林在催化剂,优选均相酸(无机或有机)和有机胺存在下在大于2巴的升高的绝对压力下制备甲基丙烯醛。实例尤其描述在ep 2 998 284 a1或us 7,141,702、jp 3069420、jp 4173757、ep 0 317 909或us 2,848,499中。根据de 32113681的方法也合适,其中以数秒范围的停留时间实现高转化率和收率。但是,后一公开文本也提到了二聚甲基丙烯醛("dimal")的产生,这最终构成损失。这些方法在升高的温度和压力下进行,优点是反应物的停留时间短和因此反应器容积较小。
[0058]
b.us 4,408,079是与上述方法相对照在较低温度和长得多的停留时间下运行的方法的一个实例,其因此需要较高的反应器容积。这些方法在2巴以上的绝对压力下进行。但是,同时,需要量大得多的催化剂
–
在一些情况下甚至50摩尔%,但在此催化剂溶液也可再循环。通常,在这些方法变化方案中,使用搅拌或循环泵送的搅拌釜或所述釜的级联。这种方法变化方案中特征是作为副产物的较高dimal含量,这归因于较长停留时间和因此提高的甲基丙烯醛的diels-alder反应的发生率。
[0059]
c.用于制备甲基丙烯醛的第三种方法变化方案的特征在于异丁烯或叔丁醇在气相中在非均相催化剂上与水蒸气和含氧的气体在300℃以上的温度下反应,然后分离。在相关现有技术中描述了许多子变化方案和可用的催化剂体系和分离选项。下列参考文献给出
了这方面的很好的综述:trends and future of monomer-mma technologies,k.nagai&t.ui,sumitomo chemical co.,ltd.,basic chemicals research laboratory,2005,http://ww.sumitomo chem.co.jp/english/rd/report/theses/docs/20040200_30a.pdf。特别在亚洲地区在工业规模下实施这些使用c4原材料的方法。
[0060]
优选的是,在反应器i中的第一反应阶段是丙醛与福尔马林的反应。在这种情况下,随后进一步优选的是在第一蒸馏柱中将来自反应器ii的反应器输出物脱除甲基丙烯醛和部分地脱除醇,以在此获得包含甲基丙烯酸烷基酯、水、碱金属甲基丙烯酸盐和/或甲基丙烯酸、mibsm和醇的料流。这种料流随后在第二蒸馏中分离成包含甲基丙烯酸烷基酯和醇的轻相和包含水、mibsm、甲基丙烯酸、二聚甲基丙烯醛和任选的二聚甲基丙烯醛的烷基酯的重相。
[0061]
在这一方法变化方案中在反应器iii中二聚甲基丙烯醛可随后解离成甲基丙烯醛。在这种情况下,也有可能将任选存在的二聚甲基丙烯醛的烷基酯解离成甲基丙烯醛和与所用的醇对应的甲基丙烯酸烷基酯。在每种情况下由此获得的甲基丙烯醛可随后在后续蒸馏阶段中与甲基丙烯酸烷基酯分离并送回反应器ii。
[0062]
作为对其中反应器i中的第一反应阶段是丙醛与福尔马林的反应的所述方法变化方案的替代,该第一反应阶段也可以是叔丁醇和/或异丁烯的氧化。
[0063]
在这样的变化方案中,随后特别优选在第一蒸馏柱中将来自反应器ii的反应器输出物脱除甲基丙烯醛和部分脱除醇。由此获得包含甲基丙烯酸烷基酯、水、碱金属甲基丙烯酸盐和/或甲基丙烯酸、mibsm和醇的料流。这种料流通常首先用酸,例如无机酸,例如硫酸处理,其中中和大部分的碱金属甲基丙烯酸盐并因此形成游离甲基丙烯酸。
[0064]
这种料流随后在第二蒸馏和/或萃取中分离成包含甲基丙烯酸烷基酯和来自该反应的副产物,即大部分所形成的甲基丙烯酸、mibsm、以及dimal、dimal酯和相对少量的水和甲醇的轻相或疏水相,和重的亲水相。这种第二、主要水性的相当然含有少量有机产物并主要由水和甲醇组成,并含有来自中和过程的碱金属或碱土金属盐。
[0065]
作为本发明的一个方面,甲基丙烯醛在含氧的气体存在下、在20至150℃的中等温度下、在1至20巴的中等压力下、在非均相球形含贵金属催化剂存在下转化。多种催化剂可用于mal与甲醇的这种氧化酯化以产生mma:
[0066]
mal与甲醇直接氧化酯化以产生mma的最早已知的应用是由asahi用存在于氧化物载体上的pd-pb催化剂进行的。us 6,040,472描述了这些催化剂,但这些相比而言仅以不足的活性和选择性产生mma。在这种情况下,催化剂是具有壳结构的含pd/pb的催化剂。对mma的选择性据报道最多为91%,且时空收率据报道最多为5.3mol。此外,在此,铅掺杂对活性氧化物类的形成至关重要,但通过铅离子的渐进的损失造成上述缺点。这些催化剂的副产物,也如在所有其它体系中那样,是甲基丙烯酸和其它副产物。
[0067]
ep 1 393 800描述了含金的催化剂,其中被描述为活性氧化物类的催化性金粒子必须尤其具有小于6nm的平均直径。所述金粒子分布在氧化硅载体或tio2/sio2载体上。作为除金外的附加活性组分,这样的催化剂尤其还含有氧化物形式的其它金属。协同效果和提高活性和选择性的效果归因于这些掺杂组分。
[0068]
通过将金盐和其它金属盐施加到氧化物载体上来进行制备。
[0069]
haruta等人在j.catal.1993,第144卷,第175-192页中公开了,施加到过渡金属氧
化物载体,如tio2、fe2o3或co3o4上的金纳米粒子是活性氧化催化剂。在这种情况下,金和过渡金属之间的相互作用对催化剂活性起到关键作用。
[0070]
ep 2 177 267和ep 2 210 664描述了具有壳结构的含镍催化剂。在这些催化剂的情况下对mma的选择性最多为97%。在该催化剂中大约1重量%的金含量下,时空收率据描述为9.7mol mma/(kg h)。根据实施例,nio
x
/au催化剂表现出好得多的活性和对mma的选择性,而其它组合,例如au与cuo或还有co3o4,活性和选择性低得多。原则上,这是上述催化剂的进一步发展,其中金-ni氧化物复合粒子的不均匀分布和非活性催化剂壳是这些新催化剂的特征。在这些中也提到作为副产物形成甲基丙烯酸。
[0071]
ep 2 210 664公开了在所谓蛋壳结构的形式的外部区域中具有在由sio2、al2o3和碱性元素,尤其是碱金属或碱土金属组成的载体上的氧化镍和金纳米粒子的催化剂。氧化镍在此富集在表面处,但也以较低浓度存在于催化剂粒子的更深层中。
[0072]
wo 2017/084969 a1描述了基于两种或更多种混合氧化物作为载体的催化剂体系,其同样包括纳米微粒状的金以及氧化钴作为活性组分。催化活性组分,即金和钴的分布是在颗粒的横截面上的各向异性分布。在us 9,676,699中描述了用于所述反应的另外更新的催化剂。在此描述了基于二氧化硅、氧化铝、碱土金属氧化物混合物的类似载体体系,其包括钯以及铋,以及各种不同的第三掺杂剂。在此也提到水浓度和作为副产物形成的甲基丙烯酸之间的相关性。
[0073]
因此,这些催化剂体系在所用载体材料方面不同,同样其制备以及最终在转化率和选择性方面的性能也不同。尽管如此,所有这些催化剂体系带来类似的副产物特征。所有催化剂的共同点在于,在稳态运行中,除所需甲基丙烯酸烷基酯外,还形成或多或少的量的甲基丙烯酸、烷氧基异丁酸酯(aibsm)和所用甲基丙烯醛的二聚物和这些二聚物的烷基酯。另外形成其它副产物,如羟基异丁酸及其相应的酯。这些副产物是特别相关的,因为它们相对于所需甲基丙烯酸烷基酯是高沸点的,并在后处理过程中富集和最终收集在相对于mma的高沸点馏分中。
附图说明
[0074]
图1的附图标记列表
[0075]
图1是根据本发明的mma方法的可能的流程图的一个实例,其包括用于由mibsm和mas获得mma的反应器
[0076]
(1)用于mal合成的反应器i
[0077]
(2)蒸馏柱
[0078]
(3)用于doe反应的反应器ii
[0079]
(4)mal分离出
[0080]
(5)中间柱和/或萃取
[0081]
(6)用于甲醇分离脱除的柱
[0082]
(7)用于mma提纯的柱
–
高沸物
[0083]
(8)用于mma提纯的第二柱
–
低沸物
[0084]
(9)用于mma提纯的第三柱
–
提纯柱
[0085]
(10)纯化的mma
[0086]
(11)任选的用于减少来自(7)的柱底物料流中的mma的量的柱
[0087]
(12)为(13)加入酸和meoh
[0088]
(13)用于将mibsm解离成mma和将dimal酯解离成mal和mma和将mas酯化成mma的反应器iii
[0089]
(14)任选的用于将有价值的产物与高沸物&硫酸分离的柱
[0090]
(15)废水
[0091]
(16)甲基丙烯醛和甲醇的再循环
具体实施方式
[0092]
实验部分:
[0093]
实施例1:用新鲜反应物mas和mibsm作为进料批料在标准压力下进行反应
[0094]
在带有安装上的柱的玻璃制三颈烧瓶中进行该反应。
[0095]
三颈烧饼配有精密玻璃(kpg)搅拌器和具有40mm的净直径的1m高的柱;借助油浴进行加热。该柱装有拉西环;在柱段的顶部安置回流分配器以能够控制回流和取出。向1升三颈烧瓶初始装载2摩尔mibsm和2摩尔甲基丙烯酸和0.2摩尔水。
[0096]
在每种情况下向这种混合物中加入200ppm的吩噻嗪和50ppm的tempol作为稳定剂并用于抑制(甲基)丙烯酸系反应物和产物在反应条件下的任何自由基聚合。借助油浴将反应混合物加热到150℃;在10分钟后,用预热油浴达到这一温度;将柱切换到完全回流,使得在一开始没有产生馏出物。在达到150℃的内部温度后,经由浸入的毛细管将mibsm、mas和meoh、水和硫酸的混合物以150g/h的计量速率连续进给到反应混合物中。借助hplc泵计量加入该进料混合物;一旦该反应运行到稳态,第二hplc泵经由毛细管输出生成的反应柱底物。
[0097]
反应物和催化剂、醇和水分开预混并经由通往搅拌器下方的毛细管引入该反应。进料混合物的组成:
[0098]
表1:用于mibsm解离成mma并且同时mas平行酯化成mma的进料混合物
[0099][0100]
因此,c4副产物mas和mibsm的摩尔比为1:1。水含量为基于mibsm计49摩尔%,和基于mibsm计142摩尔%meoh。
[0101]
随着进料计量的开始,将油浴加热至高温到160℃;反应器中的内部温度逐渐升高到大约150℃并在柱顶收集由meoh和mma或mma和水的二元共沸物的共沸组合物组成的混合物。一旦柱顶达到69℃的稳定温度,建立了0.8的回流比并取出馏出物。
[0102]
该反应最初连续运行6小时,其中在每种情况下每小时量化和分析馏出物的量。运行该系统以使平均每小时供入的反应物质量的大约90%在柱顶作为馏出物取出,同时同样
连续排出反应柱底物;借助hplc泵除去供入的进料料流的平均大约10%。平均而言,由此保持烧瓶中的料位并且该反应在这一状态下在体积或质量方面可被视为稳态。在柱顶部,在用具有大约18℃的冷却水温度的自来水运行的冷凝器下游连接安装冷阱以捕集挥发性组分;冷阱用丙酮/干冰的混合物在接近-60℃下运行;冷阱装有thf以吸收和定性表明和测定挥发性组分。
[0103]
柱底物最初变黄,然后在6小时内变浅橙色;几乎没有察觉到粘度的任何升高。
[0104]
在稳态下作为馏出物获得的柱顶产物重量为134.9g/h并通过gc色谱法具有下列组成:
[0105]
表2:该反应的馏出物产物
[0106][0107]
在稳态下获自排料的底部产物重量为15.1g/h并具有下列组成:
[0108]
表3:来自该反应的底部产物
[0109][0110]
根据供入的反应物的量,这相当于基于mibsm计96%、基于酯化成mma的甲基丙烯酸计95%的mma计算收率和93%的甲醇回收率。
[0111]
在冷阱中,除用作吸收剂的thf溶剂外,借助气相色谱法还可检测到少量二甲醚(沸点-24℃)。
[0112]
该实验表明,在所选条件下,可在化学计算量的硫酸存在下和在meoh和水存在下使用包含mas和mibsm的混合物以高效率和高转化率(基于反应物计)制备粗制mma。
[0113]
实施例2至9:
[0114]
由丙醛和福尔马林制备甲基丙烯醛:根据ep 2 998 284制备和分离甲基丙烯醛。
[0115]
借助静态混合器混合根据实施例而定具有37重量%或55重量%的福尔马林含量
的福尔马林溶液和丙醛(下文称为醛溶液),随后在用油加热的热交换器中将该混合物加热到所需温度(见表1)。根据实施例而定的福尔马林的确切水含量没有进一步的作用,因为这完全进入根据表1的新鲜进料的水含量中。从与管式反应器连接的产物柱底部的再循环料流与乙酸和二甲胺(作为40%水溶液)混合并同样预热到所需温度。预热的醛溶液和预热的催化剂溶液在另一静态混合器中混合。然后将这种反应物混合物供入借助油调温的管式反应器。该反应通常在大约35至40巴的压力下进行。
[0116]
管式反应器流出的产物混合物经阀释放并进入用于蒸馏的产物柱。在该柱的顶部,在冷凝和相分离后,获得甲基丙烯醛和水相的两相混合物。将水相送回该柱。有机相进入产物接收容器。在该柱的底部,将一个分料流作为再循环物送回该反应。将另一分料流作为水性产物排出到另一产物接收容器中。在实施例1至4中,获得具有小于0.2重量%的dimal含量的甲基丙烯醛品质。水含量为大约56重量%且基于进料中的水计的二甲胺含量为大约2.7重量%。反应器中的温度在作为入口温度的122℃和作为出口温度的153℃之间。没有出现明显的温度峰。
[0117]
实施例5至7表明,反应方案的参数对转化率和dimal含量具有至关重要的影响,因为在此可实现低于0.4重量%,但不低于0.2重量%的二聚mal含量。与实施例1至4的区别在此除了在一些情况下更高的入口温度以外,特别是更高的最大温度和出口温度。
[0118]
实施例8和9显示产生具有低于0.5重量%的二聚mal含量的甲基丙烯醛品质的实施方案。在此,入口温度和特别地,最大温度更高。
[0119]
更特别地,最大温度高于165℃或甚至170℃的优选最大温度。
[0120][0121]
如上所述制备的甲基丙烯醛在反应后卸压(任选在膨胀箱(flashbox)中部分蒸发)并导入蒸馏柱。在蒸馏柱顶部,在冷凝后,获得两相混合物(根据温度,分离出或多或少
的水相),其中上层相含有>97%的品质的甲基丙烯醛,水含量为1-3重量%。该甲基丙烯醛中的福尔马林含量<2000ppm;甲醇含量,取决于所用福尔马林的甲醇含量,在0.1至1.0重量%之间。根据上述实施例,该甲基丙烯醛含有0.18重量%至<1重量%的dimal含量。这一品质在以下实验中用于与甲醇的直接氧化酯化。
[0122]
实施例10:在液相中的直接氧化酯化的实施
[0123]
在带有鼓泡搅拌器的20升反应器中,在8重量%催化剂的浆料密度下装载由在甲醇中的36重量%甲基丙烯醛组成的反应混合物。在80℃下搅拌的同时使反应混合物达到5巴,并计量加入空气以使冷凝器下游的尾气中的氧浓度为4.0体积%。通过连续引入4重量%naoh/甲醇溶液,将ph值调节到7。从反应器中连续排出反应混合物,其形式使得催化剂装载量为11mol mal/kg催化剂/小时。运行时间在每种情况下为1000小时。
[0124]
a)所用催化剂是金-钴氧化物催化剂(wo2017084969 a1)并在94.1%mma选择性下获得76%mal的转化率。对mas的选择性为3.1%且对mibsm的选择性为1.2%。
[0125]
b)所用催化剂是金-镍氧化物催化剂(us8450235)并在94.4%mma选择性下获得75%mal的转化率。对mas的选择性为2.5%且对mibsm的选择性为1.2%。
[0126]
c)所用催化剂是钯-铅催化剂(us5969178),其中将该反应的ph值调节到6.3,并在89%mma选择性下获得60%mal的转化率。对mas的选择性为7%且对mibsm的选择性低于0.1%。
[0127]
d)所用催化剂是钯-铋-碲催化剂(us20160168072)并根据如us20160168072中所述的实施例2和3设定条件和化学计量。在92%mma选择性下获得89%的转化率。对mas的选择性低于0.2%且对mibsm的选择性为1.2%。
[0128]
实施例11:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。硫酸作为催化剂
[0129]
获自实施例3a的反应混合物在后处理后提取:
[0130]
举例描述该后处理,其中各自的组成列在表1中。
[0131]
将来自反应器ii的输出物(1000g/hr)在柱级22中的第11级/处导入mal回收柱。柱底温度为70℃,在930毫巴的压力下。柱底料流用硫酸酸化至ph 2,在滗析器中分离,并将有机相送入萃取柱的底部,其中将水相引入30级萃取柱的顶部。萃取柱的柱底温度为43.9℃,在1013毫巴的压力下。将来自萃取柱的顶部料流引入高沸物柱的柱级10中的第6级。柱底温度为85.4℃,在235毫巴的压力下。
[0132]
表5:在各不同后处理步骤中的反应混合物的组成
[0133][0134]
收集来自高沸物柱的底部产物并用于将mibsm解离成mma和meoh,和将dimal酯解离成mal和mma,同时用meoh将mas酯化以产生mma。
[0135]
500毫升三颈烧瓶配有柱和玻璃温度计。在柱顶部,在滴液漏斗中加入50克含吩噻嗪(大约500ppm)的meoh,以通过连续添加防止在该柱中聚合。将热电偶置于油浴中(t(油)=165℃)。
[0136]
将300克进料(1当量,0.73mol 3-mibsm;1.17当量,0.85mol mas;0.34mol mma,0.23mol dimal酯),
[0137]
2.84克(0.04当量,0.029mol)h2so4和
[0138]
13.42克h2o(1.02当量、0.75mol)
[0139]
初始装载在置于油浴(额定温度=165℃)中的三颈烧瓶中。
[0140]
将该混合物加热3小时到165℃(油浴额定温度),其中达到151℃的底部温度。连续取出馏出物并通过hplc分析。在3小时的反应过程中,加入甲醇-稳定剂溶液(6.54g/h,0.20mol)。
[0141]
下表2显示所用硫酸、水、甲醇和进料样品的量。也列出进料样品的3-mibsm、mas和mma的组成以及摩尔质量。
[0142]
表6:所用物质的量和它们的摩尔质量
[0143][0144]
表7显示所用的上文所列反应物的质量回收率。
[0145]
表7:质量平衡的结论
[0146][0147]
该质量平衡在98.90%的回收率。表8显示馏出物和柱底物中的组分的摩尔分布。
[0148]
表8:馏出物和柱底物中的组分的分布
[0149][0150]
该实验成功;3-mibsm和mas转化成mma的转化率高。
[0151]
发现96.0%的3-mibsm转化成mma且87.3%的mas转化成mma。mas和3-mibsm对mma的选择性合计为99.16%。dimal酯热解离成mal和mma的转化率为8.26%。
[0152]
实施例12:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。磷酸作为催化剂
[0153]
该反应类似于实施例11进行并使用磷酸代替硫酸。
[0154]
该实验成功;3-mibsm和mas转化成mma的转化率高。
[0155]
发现94.2%的3-mibsm转化成mma且86.8%的mas转化成mma。mas和3-mibsm对mma的选择性合计为99.0%。dimal酯热解离成mal和mma的转化率为8.25%。
[0156]
实施例13:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。甲磺酸作为催化剂
[0157]
该反应类似于实施例11进行并使用甲磺酸代替硫酸。
[0158]
该实验成功;3-mibsm和mas转化成mma的转化率高。
[0159]
发现92.6%的3-mibsm转化成mma且84.7%的mas转化成mma。mas和3-mibsm对mma的选择性合计为98.8%。dimal酯热解离成mal和mma的转化率为8.20%。
[0160]
实施例14:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。温度为120℃
[0161]
该反应在120℃的温度下类似于实施例11进行。
[0162]
3-mibsm和mas转化成mma的转化率高。
[0163]
发现60.4%的3-mibsm转化成mma且87.3%的mas转化成mma。dimal酯热解离成mal和mma的转化率为8.20%。
[0164]
实施例15:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。温度为90℃
[0165]
该反应在90℃的温度下类似于实施例11进行。
[0166]
3-mibsm到mma的转化率不高,但mas到mma的转化率高。
[0167]
发现小于1%3-mibsm转化成mma且88.0%的mas转化成mma。dimal酯没有热解离成mal和mma。
[0168]
实施例16:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。增加的硫酸的量
[0169]
该反应类似于实施例11进行,使用40摩尔%的增加的硫酸量。
[0170]
该实验成功;3-mibsm和mas转化成mma的转化率高。
[0171]
发现95.9%的3-mibsm转化成mma且87.4%的mas转化成mma。mas和3-mibsm对mma的选择性合计为99.14%。dimal酯热解离成mal和mma的转化率为8.16%。
[0172]
对比例1:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。温度为23℃
[0173]
该反应在23℃的温度下类似于实施例11进行。
[0174]
3-mibsm到mma的转化率和mas到mma的转化率不高。
[0175]
发现小于1%3-mibsm转化成mma且20%的mas转化成mma。dimal酯没有热解离成mal和mma。
[0176]
对比例2:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm解离成mma和甲醇并同时将mas酯化成mma。没有加入硫酸作为催化剂
[0177]
该反应类似于实施例11进行,但没有加入硫酸为催化剂。
[0178]
3-mibsm到mma的转化率高,但mas没有转化成mma。
[0179]
发现94%的3-mibsm转化成mma且mas没有转化成mma。dimal酯热解离成mal和mma的比率为8.0%。
[0180]
实施例17:用来自实施例3的反应混合物在根据本发明分离出甲基丙烯醛和甲基丙烯酸甲酯后将mibsm连续解离成mma和甲醇并同时将mas酯化成mma。
[0181]
该反应类似于实施例11进行。此外,在达到151℃的底部温度后,开始连续进给57g/hr的进料混合物。该实验运行6小时。
[0182]
该实验成功;3-mibsm和mas转化成mma的转化率高。
[0183]
发现95.5%的3-mibsm转化成mma且87.5%的mas转化成mma。mas和3-mibsm对mma的选择性合计为98.7%。dimal酯热解离成mal和mma的转化率为8.05%。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips