
 商标分类
商标分类  商标转让
商标转让 
一种异硫氰酸酯类液晶单体的合成方法与流程
 2021-02-02 04:02:46|
2021-02-02 04:02:46| 404|
404| 起点商标网
起点商标网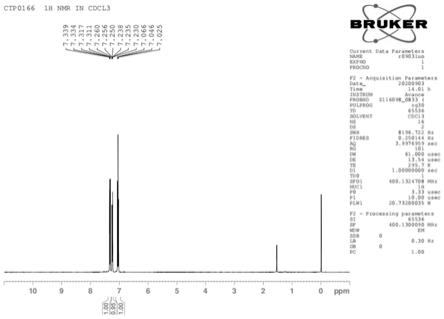
[0001]
本发明属于液晶材料技术领域;涉及一种液晶单体的合成方法;更具体地,涉及一种异硫氰酸酯类液晶单体的合成方法。
背景技术:
[0002]
近年来随着触控屏幕等广电显示材料的普及,液晶作为其中必不可少的部分受到了格外的青睐及发展应用,随着对液晶材料的研究发现,近年来,液晶器件逐渐应用在光束偏转器、可调谐棱镜、场序显示器、空间光调制器、红外场景生成器等领域,在这些光学应用中,往往对液晶材料的光学性质要求较高,研究发现,异硫氰酸酯(ncs)液晶具有较高双折射率δn和适中的旋转黏度,响应性能fom值通常较大,适合用来制作成液晶光学器件。异硫氰酸酯类液晶材料因为其良好的物理化学稳定性、较宽的工作温度范围、适当的低黏度,以及快速响应与低电压驱动等优点,故而在光电显示材料有着更加广泛的应用空间和研究价值。
[0003][0004]
其中r1、r2=h、f;r3=ch3、ch3ch2、ch3ch2ch2……
[0005]
有些异硫氰酸酯类产品在对产品结构上的忽略使得合成路线不是最优,最终产品或不可得到。以炔基型异硫氰酸酯类液晶材料为例,在一些公开的文献中显示虽然可制得目标产物但收率较低,且相对处理方式较为复杂,因此本专利方法在这些合成反应和后处理方面上进行了优化,在保证产品纯度下也实现了较高的收率。
[0006]
在本专利中我们设计了一种活性中间体结构如下图所示,当以此中间体为底物可较为简便的制备出上文中提及的异硫氰酸酯类化合物,当与不同结构的偶联底物反应时能衍生出更多种类的异硫氰酸酯类液晶材料。但值得关注的是当r2、r3不全为f,r1为i的条件下任然能保持较高的活性与碘发生c-c键的偶联反应。该产品活性按着i、br、cl的反应活性逐渐降低,产品也较难提取。但仍能得到对应的异硫氰酸酯类化合物。
[0007][0008]
其中r1=cl、br、i;r2、r3=h、f。在实际生产过程中,从经济性原则出发,我们优选了以下几个反应实例以供参考。
技术实现要素:
[0009]
本发明的目的之一在于提供一种异硫氰酸酯类液晶单体的合成方法。所述合成方
法先制备异硫氰酸酯基的活性中间体,再与不同的偶联底物反应后再经提纯得到一系列异硫氰酸酯类液晶单体。
[0010]
为实现上述目的,本发明采用如下技术方案:一种异硫氰酸酯类液晶单体的合成方法,以卤代苯胺为原料,经反应和提纯最终得到异硫氰酸酯类液晶单体;其特征在于,所述反应包括如下步骤:
[0011]
(1)成盐:以乙醇为溶剂,卤代苯胺溶液投入cs2、三乙烯二胺;过夜反应,得到盐;
[0012]
(2)异硫氰酸酯化:以dcm为溶剂,盐在低温下滴加dcm-三光气溶液,得到活性中间体;
[0013]
(3)偶联:投入钯催化剂后n2置换釜内空气,活性中间体和偶联底物在碱作用下在60-100℃下回流反应2-48h。
[0014]
根据本发明所述的合成方法,其中,在成盐反应中,卤代苯胺:cs2:三乙烯二胺的摩尔比=1:3~5:3~5。
[0015]
根据本发明所述的合成方法,其中,在成盐反应中,乙醇用量为卤代苯胺的 5~10倍体积。
[0016]
根据本发明所述的合成方法,其中,在异硫氰酸酯化反应中,盐:三光气的摩尔比=1:0.33~0.5。
[0017]
有利地,低温表示温度-5-5℃之间。
[0018]
根据本发明所述的合成方法,其中,异硫氰酸酯化或也可经硫光气直接制备,具体配比为:盐:硫光气的摩尔比=1:3~5。
[0019]
根据本发明所述的合成方法,其中,在异硫氰酸酯化反应中,dcm用量为卤代苯胺的5~10倍体积。
[0020]
根据本发明所述的合成方法,其中,在偶联反应中,活性中间体:偶联底物:碱的摩尔比=1:1~1.2:2~4。
[0021]
根据本发明所述的合成方法,其中,所述钯催化剂选自0价钯催化剂或二价钯催化剂。
[0022]
根据本发明所述的合成方法,其中,所述钯催化剂选自四三苯基膦钯或双三苯基二氯化钯。
[0023]
根据本发明所述的合成方法,其中,所述钯催化剂用量为卤代苯的 0.1mol%~0.01mol%。
[0024]
根据本发明所述的合成方法,其中,所述偶联底物选自对甲苯硼酸和对正丙基苯乙炔。
[0025]
本专利在这些合成反应和后处理方面上进行了优化,在保证产品纯度下也实现了较高的收率。
附图说明
[0026]
图1:活性中间体:2-氟-4-溴-1-异硫氰酸酯基苯gc图谱。
[0027]
图2:活性中间体:2-氟-4-溴-1-异硫氰酸酯基苯hnmr图谱。
[0028]
图3:8-甲基-2-氟-1-异硫氰酸酯基联苯gc图谱。
[0029]
图4:8-甲基-2-氟-1-异硫氰酸酯基联苯hnmr图谱。
[0030]
图5:1,3-二氟-2-异硫氰酸根合5-[2-(4-丙基苯基)乙炔基]苯gc图谱。
[0031]
图6:1,3-二氟-2-异硫氰酸根合5-[2-(4-丙基苯基)乙炔基]苯hnmr图谱。
具体实施方式
[0032]
下面结合实施例,进一步说明本发明,并不限定本发明的应用。除非另有说明,实施例中的百分数一律是质量百分数。
[0033]
实施例1:单f联苯型异硫氰酸酯类液晶单体
[0034]
活性中间体的制备
[0035]
成盐:在装有机械搅拌的1l的三口烧瓶中,投入2-氟-4-溴苯胺50g、乙醇 500ml,开启搅拌使产品完全溶解后,再加入二硫化碳60g及三乙烯二胺88.63g,室温搅拌过夜(15h左右),tlc分析检测无2-氟-4-溴苯胺即认为到达反应终点进行后处理。
[0036]
后处理:反应液过滤的固体即为产品粗品。再用理论质量体积的2-3倍乙醇进行淋洗可得淡黄色固体,烘干后即可直接投入下一步反应中。
[0037]
异硫氰酸酯化:在装有机械搅拌的2l的三口烧瓶中,投入盐99g、二氯甲烷(dcm)1l,开启搅拌使产品混合均匀后,n2保护下1h内缓慢滴加dcm
-ꢀ
三光气溶液(31.06g三光气溶于200mldcm),过程控制温度-5-5℃之间,滴加完毕后,在0-5℃保温1h,随即转移至40℃下回流搅拌2-3h;降温至室温后,加60ml水猝灭三光气,搅拌5min使反应液呈透明无明显固体存在(若仍有部分固体附着在壁上,补加水使之完全溶解)。
[0038]
后处理:反应液加水后静置分液,有机相干燥后,旋去溶剂,并用石油醚(pe) 带三次,使dcm完全除去,旋干得粗品,称重;粗品完全溶解于5倍体积的热 pe中,过热柱(快速柱),旋干pe可得白色产品53.03g。
[0039]
送检hplc/gc,纯度99.23%,收率:87.33%。
[0040]
活性中间体检测结果见附图1-2。
[0041]
偶联:在装有磁子的500ml的三口烧瓶中,投入活性中间体20g、对甲基苯硼酸12.25g、乙醇200ml,加入水40ml、碳酸钾23.8g,开启搅拌,最后投入pd催化剂0.053g(四三苯基膦钯),置换釜内空气,n2保护下80℃回流过夜 (15h左右),tlc分析检测无活性中间体时即认为到达反应终点进行后处理。
[0042]
后处理:硅藻土过滤后,旋蒸除去乙醇,加水、dcm分液,收集有机相,再用0.5mol/l50ml稀盐酸洗涤两次后分液,有机相干燥后旋蒸除去dcm得棕褐色固体,再经层析柱分离得白色产物13.78g。
[0043]
最终得到产品纯度99.5%,收率65.18%。检测结果见图3-4。
[0044]
实施例2:双f联苯型异硫氰酸酯类
[0045]
活性中间体的制备
[0046]
成盐:在装有机械搅拌的1l的三口烧瓶中,投入2,6-二氟-4-溴苯胺50g、乙醇500ml,开启搅拌使产品完全溶解后,再加入二硫化碳54.85g及三乙烯二胺80.81g,室温搅拌过夜(15h左右),tlc分析检测无2,6-二氟-4-溴苯胺即认为到达反应终点进行后处理。
[0047]
后处理:反应液过滤的固体即为产品粗品。再用理论质量体积的2-3倍乙醇进行淋洗可得淡黄色固体,烘干后即可直接投入下一步反应中。
[0048]
异硫氰酸酯化:在装有机械搅拌的2l的三口烧瓶中,投入盐93g、 dcm900ml,开启
搅拌使产品混合均匀后,n2保护下1h内缓慢滴加dcm-三光气溶液(27.85g三光气溶于250mldcm),过程控制温度-5-5℃之间,滴加完毕后,在0-5℃保温1h,随即转移至40℃下回流搅拌2-3h;降温至室温后,加60ml 水猝灭三光气,搅拌5min使反应液呈透明无明显固体存在(若仍有部分固体附着在壁上,补加水使之完全溶解)。
[0049]
后处理:反应液加水后静置分液,有机相干燥后,旋去溶剂,并用pe带三次,使dcm完全除去,旋干得粗品,称重;粗品完全溶解于5倍体积的热pe中,过热柱(快速柱),旋干pe可得白色产品48.31g。
[0050]
送检hplc/gc,纯度98.93%,收率:82.33%。
[0051]
偶联:在装有磁子的250ml的三口烧瓶中,投入活性中间体10g、对甲基苯硼酸5.70g、乙醇100ml,加入水20ml、碳酸钾11g,开启搅拌,最后投入 pd催化剂0.024g(四三苯基膦钯),置换釜内空气,n2保护下80℃回流过夜(15h 左右),tlc分析检测无活性中间体时即认为到达反应终点进行后处理。
[0052]
后处理:硅藻土过滤后,旋蒸除去乙醇,加水、dcm分液,收集有机相,再用0.5mol/l 25ml稀盐酸洗涤两次后分液,有机相干燥后旋蒸除去dcm得棕褐色固体,再经层析柱分离得白色产物5.78g。
[0053]
最终得到产品纯度99.5%,收率55.37%。
[0054]
实施例3:双f炔基苯型异硫氰酸酯类
[0055]
活性中间体的制备
[0056]
异硫氰酸酯化:在装有磁力搅拌的250ml的三口烧瓶中,投入2,6-二氟-4
-ꢀ
溴苯胺10g、dcm100ml,开启搅拌使产品完全溶解后,再加入碳酸钠溶液(16g 碳酸钠加入10ml水),置于冰水浴中,当体系温度小于5℃时开始缓慢滴加硫光气溶液30ml(30%),滴加完毕后0-5℃下保温1h再自然升温至室温保温两小时,经tlc监控无原料剩余即可停止反应进行后处理。
[0057]
后处理:向反应液中补加等量碳酸钠溶液猝灭硫光气后,硅藻土过滤,分液。有机相干燥后旋干溶剂,粗品完全溶于两倍体积的乙醇中后,冷却析晶过滤的粗品,经硅胶柱分离后的白色粉末固体10.12g。
[0058]
偶联:在装有磁子的100ml的三口烧瓶中,投入活性中间体10g、三乙胺 50ml,对正丙基苯乙炔6.34g、碘化亚铜76.16mg、三苯基膦0.209g,最后加入钯催化剂0.56g,n2置换釜内空气。在回流温度下搅拌过夜(15h左右),tlc 分析检测无原料即认为到达反应终点进行后处理。
[0059]
后处理:硅藻土过滤后,旋蒸除去乙醇,加水、dcm分液,收集有机相,再用0.5mol/l50ml稀盐酸洗涤两次后分液,有机相干燥后旋蒸除去dcm得棕褐色固体,再经层析柱分离得白色产物4.21g。
[0060]
最终产品纯度:98.82%,收率:33.60%。检测结果见图5-6。
[0061]
应理解,本发明的具体实施方式仅用于阐释本发明的精神和原则,而不用于限制本发明的范围。此外应理解,在阅读了本发明的内容之后,本领域技术人员可以对本发明的技术方案作出各种改动、替换、删减、修正或调整,这些等价技术方案同样落于本发明权利要求书所限定的范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips