
 商标分类
商标分类  商标转让
商标转让 
一种酰基膦光引发剂及其制备方法与流程
 2021-02-02 03:02:04|
2021-02-02 03:02:04| 293|
293| 起点商标网
起点商标网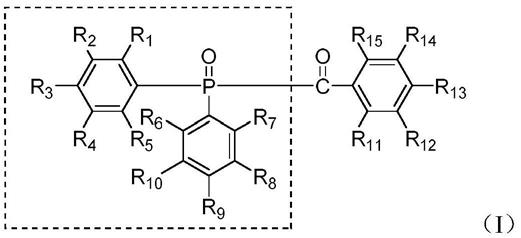
[0001]
本发明属于有机合成及应用技术领域,具体涉及一种酰基膦光引发剂及其制备方法。
背景技术:
[0002]
光固化技术与传统的固化技术相比,拥有高效、节能、环保、经济、适用性广等特点,越来越多的应用于涂料、油墨、胶黏剂的固化。光固化体系一般包括:光源、单体和引发剂,其中光源与引发剂波长的匹配性决定着固化配方的优劣。与uv光引发相比,近uv光和可见光引发更加安全、节能,同时长波长的光可适用于厚涂层的固化,因此开发与近uv光和可见光相匹配的长波长引发剂是光固化领域的研究热点之一。
[0003]
2,4,6-三甲基苯甲酰基-二苯基氧化膦(tpo,如式v)是一款商业化的适用于涂层深层固化的光引发剂,其传统的制备方法有两种:(1)酰氯法。其制备过程是苯基二氯化磷(bpd)高温歧化制得二苯基氯化磷(dpc),二苯基氯化磷酯化后与均三苯甲酰氯反应得到产品,如反应式1;(2)醛法。其制备过程是bpd高温歧化制得dpc,dpc 水解得到二苯基氧化膦(dpo),dpo与均三甲基苯甲醛反应后经氧化得到产品,如反应式2;这两种制备tpo的方法都包括bpd的高温歧化制备dpc过程,在这个过程中产生大量的催化剂固废,严重污染环境,因此开发tpo绿色环保的合成路线是研究热点之一。
[0004][0005]
反应式1酰氯法制备tpo
[0006][0007]
反应式2醛法制备tpo
[0008]
为了改善tpo的引发效率,大量的文献报道了对tpo进行结构改性,但目前为止,对tpo进行结构改性的研究主要集中在对苯甲酰基中苯环结构进行修饰(式v中右侧框内结构),关于膦酰基中苯环结构(式v中左侧框内结构)修饰的研究还未见报道。进一步的,当在
tpo的结构中引进表面能较小的氟原子时能促使引发剂在固化体系中存在上浮现象,这类引发剂在固化体系中产生梯度分布,引发剂在固化体系表面富集,能大大增强表层固化效果,降低氧阻问题。
技术实现要素:
[0009]
本发明的目的在于提供一种酰基膦光引发剂,并提供此化合物的制备方法及应用。
[0010]
本发明所采用的技术解决方案是:
[0011]
一种酰基膦光引发剂,其特征在于:酰基膦光引发剂的化学结构式如(i)所示:
[0012][0013]
其中r
1-r
15
独立的选自h、烷基、卤素、卤素取代的烷基、芳基,其中卤素包括:f、 cl、br、i,优选f。
[0014]
进一步的,所述烷基、卤素取代的烷基优选碳原子数1-6的碳链结构。
[0015]
上述酰基膦光引发剂的制备方法,包括以下步骤:
[0016]
(1)将芳基二氯化膦或其衍生物、芳烃或其衍生物、催化剂投入到反应瓶中,升温到一定温度,反应一定时间,反应完成后减压蒸出未反应的原料,得到二芳基氯化膦与催化剂的络合物;
[0017]
(2)在一定温度下,将所得二芳基氯化膦与催化剂的络合物缓慢滴加到水中,滴加完成后,继续反应一定时间,加入萃取剂萃取,有机层分出待用;
[0018]
(3)将所得的有机层投入反应瓶中,在-10~25℃下投入芳醛或其衍生物,反应 0.5~5h,反应液待用;
[0019]
(4)在一定温度下,将催化剂、氧化剂投入步骤(3)所得反应液中,反应一定时间,制得酰基膦光引发剂;
[0020]
上述过程中第一步所用芳基二氯化膦或其衍生物的化学结构式如(ii)所示:
[0021][0022]
其中r
1-r5独立的选自h、烷基、卤素、卤素取代的烷基、芳基,其中卤素包括:f、 cl、br、i,优选f。
[0023]
所述烷基、卤素取代的烷基优选碳原子数1-6的碳链结构。
[0024]
上述过程中第一步所用芳烃或其衍生物化学结构式如(iii)所示:
[0025][0026]
其中r
6-r
10
独立的选自h、烷基、卤素、卤素取代的烷基,其中卤素包括:f、cl、br、 i,优选f。
[0027]
上述过程中第三步所用芳醛或其衍生物化学结构式如(iv)所示:
[0028][0029]
其中r
11-r
15
独立的选自h、烷基、卤素、卤素取代的烷基,其中卤素包括:f、cl、 br、i,优选f。
[0030]
上述过程中第一步所用催化剂为:无水氯化铝、无水氯化镓、无水氯化铟、无水氯化铋、无水氯化锑、无水氯化锡、无水氯化镁、无水氯化铁中的一种或几种,优选:无水氯化铝;
[0031]
上述过程中第一步中一定温度指的是50~150℃,优选90℃;一定时间指的是2~48h,优选12h;
[0032]
上述过程中第一步所用芳基二氯化膦或其衍生与芳烃或衍生物物质的量之比为 1:0.5~1:5,优选1:1.3;芳基二氯化膦或其衍生物催化剂的物质的量之比为1:0.8~1:3,优选1:1.4;
[0033]
上述过程中第二步中一定的温度指的是0~50℃,优选:0℃;一定时间指的是0~20h,优选4h;所用的萃取剂选自苯、甲苯、二氯甲烷、二氯乙烷、氯仿、四氯化碳、氯苯、硝基苯中的一种或几种;
[0034]
上述过程中第三步中一定的温度指的是-10~25℃,优选:15~25℃;一定时间指的是 0.5~5h,优选1h;所用芳醛或其衍生物与芳基二氯化膦或其衍生物的物质的量之比为 1:0.6~1:1.5,优选1:1;
[0035]
上述过程中第四步中一定的温度指的是0~20℃,优选6~10℃;一定的时间指的是 1~20h,优选8h;
[0036]
上述过程中第四步中催化剂为:五氧化二钒、二乙酰丙酮合钒、二氧化锰、偏钒酸铵中的一种或几种,优选二乙酰丙酮合钒;所用催化剂为芳基二氯化膦或其衍生物物质的量的0.1%~5%,优选1%;
[0037]
上述过程中第四步中氧化剂为:高锰酸钾、双氧水、过氧化叔丁醇中的一种或几种,优选过氧化叔丁醇;所用氧化剂与芳基二氯化膦或其衍生物的物质的量之比为1:1~5:1,优选1.3:1;
[0038]
芳基二氯化膦的制备参考文献p-heteroatom-substituted arylphosphines,alajarin,m.etal,science of synthesis,31b,2105-2153;其他试剂及药品均为市售获得。
[0039]
本发明的有益技术效果是:
[0040]
(1)本发明首次对酰基膦引发剂结构中膦酰基侧的苯环进行结构修饰,制备出的酰基膦引发剂属于新结构的光引发剂;
[0041]
(2)本发明提供一类酰基膦光引发剂的制备方法,该方法具有反应条件温和、处理过程简单、绿色环保等优点,属于绿色环保工艺;
[0042]
(3)本发明制备的含f的光引发剂在固化体系中呈现梯度分布,能有效降低固化过程中固化体系表面存在的氧阻效应;制备出的含供电基团的新结构引发剂吸光能力强、吸光波长红移,大大增强了与近uv光和可见光的匹配性,作为新的引发剂具有巨大的应用潜力。
具体实施方式
[0043]
下面结合附图和具体的实施例对本发明作进一步的说明。
[0044]
图1为本发明实施例3中制得的2,4,6-三甲基苯甲酰基-2,4-二甲苯基苯基氧化膦 (2,4-dmtpo)的1hnmr谱图;1h nmr(500mhz,cdcl3)δ7.85
–
7.79(m,2h),7.68(dd, j=12.7,7.9hz,1h),7.56
–
7.51(m,1h),7.46(td,j=7.5,3.0hz,2h),7.09(dd,j=11.7, 6.0hz,2h),6.81(s,2h),2.35(d,j=4.5hz,6h),2.26(s,3h),2.06(s,6h);
[0045]
图2为本发明实施例3中制得的2,4,6-三甲基苯甲酰基-2,4-二甲苯基苯基氧化膦 (2,4-dmtpo)的
13
cnmr谱图;
13
c nmr(126mhz,cdcl3)δ219.28(d,j=71.8hz), 143.44
–
142.93(m),140.63,136.50(d,j=40.4hz),135.34,133.42
–
131.74(m),130.81(d, j=92.5hz),129.27
–
128.41(m),126.29(d,j=12.7hz),124.63(d,j=95.8hz),21.61
–ꢀ
21.18(m),19.85.
[0046]
图3为本发明实施例3中制得的2,4,6-三甲基苯甲酰基-2,4-二甲苯基苯基氧化膦 (2,4-dmtpo)的
31
pnmr谱图;
31
p nmr(202mhz,cdcl3)δ19.67;
[0047]
图4为本发明实施例4中制得的2,4,6-三甲基苯甲酰基-4-氟苯基苯基氧化膦(4-ftpo) 的1hnmr谱图;1h nmr(400mhz,cdcl3)δ8.04
–
7.95(d,j=5.5hz,4h),7.62
–
7.47(m, 3h),7.20(td,j=8.8,2.3hz,2h),6.82(s,2h),2.26(s,3h),2.03(s,6h).
[0048]
图5为本发明实施例4中制得的2,4,6-三甲基苯甲酰基-4-氟苯基苯基氧化膦(4-ftpo) 的
13
cnmr谱图;2,4,6-三甲基苯甲酰基-4-氟苯基苯基氧化膦(4-ftpo)的
13
c nmr谱图(101mhz,cdcl3)δ168.81
–
162.17(m),138.33
–
126.77(m),125.68(dd,j=96.0,3.5 hz),116.22(dd,j=21.3,12.6hz),20.49(d,j=152.5hz);
[0049]
图6为本发明实施例4中制得的2,4,6-三甲基苯甲酰基-4-氟苯基苯基氧化膦(4-ftpo) 的
31
pnmr谱图;
31
p nmr(162mhz,cdcl3)δ12.21.;
[0050]
图7为2,4,6-三甲基苯甲酰基-二苯基氧化膦(tpo),2,4,6-三甲基苯甲酰基-2,,4-二甲苯基苯基氧化膦(2,4-dmtpo)和2,4,6-三甲基苯甲酰基-4-氟苯基苯基氧化膦(4-ftpo) 的紫外吸收特征数据
[0051]
实施例1
[0052]
2,4,6-三甲基苯甲酰基-二苯基氧化膦(tpo)结构式如下
[0053][0054]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、苯基二氯化膦17.9g、苯11g 依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到二苯基氧化膦的甲苯溶液。
[0055]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的二苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到tpo。产品收率为65%,hplc 含量为99.1%。
[0056]
实施例2
[0057]
2,4,6-三甲基苯甲酰基-4-甲苯基苯基氧化膦(4-mtpo)结构式如下:
[0058][0059]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、苯基二氯化膦17.9g、甲苯12.9g 依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到4-甲苯基苯基氧化膦的甲苯溶液。
[0060]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的4-甲苯基苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到4-mtpo。产品收率为68%, hplc含量为99.1%。
[0061]
实施例3
[0062]
2,4,6-三甲基苯甲酰基-2,,4-二甲苯基苯基氧化膦(2,4-dmtpo)结构式如下:
[0063]
[0064]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、苯基二氯化膦17.9g、间二甲苯14.9g依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到2,4-二甲苯基苯基氧化膦的甲苯溶液。
[0065]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的2,4-二甲苯基苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到2,4-dmtpo。产品收率为71%,hplc含量为99.1%。
[0066]
实施例4
[0067]
2,4,6-三甲基苯甲酰基-4-氟苯基苯基氧化膦(4-ftpo)结构式如下:
[0068][0069]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、氟苯基二氯化膦17.9g、苯11g 依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到4-氟苯基苯基氧化膦的甲苯溶液。
[0070]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的4-氟苯基苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到4-ftpo。产品收率为62%, hplc含量为98.5%。
[0071]
实施例5
[0072]
2,4,6-三甲基苯甲酰基-4-三氟甲苯基苯基氧化膦(4-tftpo)结构式如下:
[0073][0074]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、4-三氟苯基二氯化膦24.7g、苯11g依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料
后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到4-三氟甲苯基苯基氧化膦的甲苯溶液。
[0075]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的4-三氟甲苯基苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到4-tftpo。产品收率为70%,hplc含量为98.5%。
[0076]
实施例6
[0077]
2,4,6-三甲基苯甲酰基-4-氟苯基-4-甲苯基氧化膦(4-f-4-mtpo)结构式如下:
[0078][0079]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、4-三氟苯基二氯化膦19.7g、甲苯13g依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到4-氟苯基-4-甲苯基氧化膦的甲苯溶液。
[0080]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的4-氟苯基-4-甲苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到4-f-4-mtpo。产品收率为67%,hplc含量为97.5%。
[0081]
实施例7
[0082]
2,4,6-三甲基苯甲酰基-4-三氟苯基-4-甲苯基氧化膦(4-tf-4-mtpo)结构式如下:
[0083][0084]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、4-三氟甲苯基二氯化膦24.7g、甲苯
13g依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到4-三氟甲苯基-4-甲苯基氧化膦的甲苯溶液。
[0085]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的4-三氟甲苯基-4-甲苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g 氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到4-tf-4-mtpo。产品收率为71%,hplc含量为98.3%。
[0086]
实施例8
[0087]
2,4,6-三甲基苯甲酰基-联苯基苯基氧化膦(dptpo)结构式如下:
[0088][0089]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(50ml)中,通入干燥的氮气,排除三口瓶中的空气。将三氯化铝17.4g、苯基二氯化膦17.9g、联苯21.7g 依次投入到三口瓶中,油浴缓慢升温至设定温度90℃,保温反应12h。减压蒸出未反应的原料后,冷却至室温;反应液缓慢滴加到100g冰水混合物中水解,水解完成后继续保温反应4h。反应液中投入120g甲苯萃取,有机层通过碱洗、水洗得到联苯基苯基氧化膦的甲苯溶液。
[0090]
在配有磁力搅拌、温度计、回流冷凝管、氮气导管的干燥的三口瓶(250ml)中,通入干燥的氮气,排除三口瓶中的空气。投入上述过程得到的联苯基苯基氧化膦、均三甲基苯甲醛14.9g在15~25℃下反应1h;降低反应液温度至6~10℃,投入0.27g氧化二乙酰丙酮合钒,缓慢滴加16.7g 70%过氧化叔丁醇,滴加完成后保温反应8h,反应液经亚硫酸钠溶液、水洗涤后蒸出溶剂,投20ml石油醚重结晶得到dptpo。产品收率为71%, hplc含量为98.1%。
[0091]
对上述实施例3和实施例4获得的产物进行测试,结果如下表:
[0092]
表1光引发剂tpo,2,4-dmtpo和4-ftpo的吸收特性数据
[0093][0094]
表2光引发剂4-ftpo固化tmpta上、下表面氟元素edx测试结果
[0095]
[0096]
说明:
[0097]
(1)与商业化的光引发剂tpo相比,本发明中制备的2,4-dmdpo引发剂最大吸收波长发生红移,更能匹配led引发光源;同时吸光系数有所增加,初步验证了其有更强的引发效果;
[0098]
(2)与商业化的光引发剂tpo相比,本发明中制备的4-ftpo最大吸收波长未发生变化,但其吸光系数有所增加,说明其具有更强的引发效果;同时因为f原子的引入,引发剂在固化体系中可发生迁移,从而产生梯度分布,该性质能够解决该类引发剂存在的表面氧阻问题。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips