
 商标分类
商标分类  商标转让
商标转让 
用于生物学衍生的甲羟戊酸转化的方法与流程
 2021-02-02 03:02:08|
2021-02-02 03:02:08| 302|
302| 起点商标网
起点商标网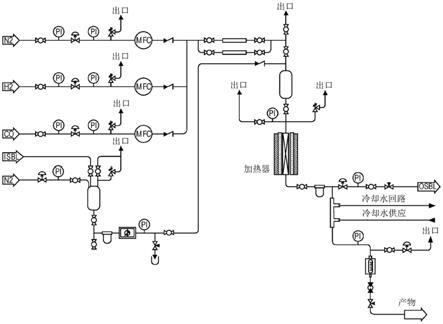
用于生物学衍生的甲羟戊酸转化的方法
[0001]
本申请是分案申请,原申请的申请日为2015年11月24日,申请号为201580062046.9,发明名称为“用于生物学衍生的甲羟戊酸转化的方法”。
[0002]
相关申请的交叉参考
[0003]
本申请要求2014年11月26日提交的题为《用于生物学衍生的甲羟戊酸转化的方法(processes for conversion of biologically derived mevalonic acid)》的美国临时专利申请第62/084,689号的优先权权益,该申请以全文引用的方式并入本文中。
[0004]
序列表
[0005]
以下申请含有在2015年11月23日创建的题为“46567序列表(46567sequencelisting)”,作为以ascii格式的25kb文本文件提交的呈计算机可读格式(crf)的序列表。crf的内容以引用的方式并入本文中。
技术领域
[0006]
本发明大体上涉及用于从通过生物化学途径衍生的有机酸产生烃生物燃料和其它产物的中间体的方法。具体来说,甲羟戊酸(或甲羟戊酸内酯)可衍生自生物质发酵以产生多种中间体。本发明的一个或多个实施例涉及异戊二烯、甲基乙烯基酮、3-甲基-2-丁酮和脱水-甲羟戊酸内酯的产生。
背景技术:
[0007]
有机酸是一类重要的化合物,它们可通过生物化学途径衍生并且可充当用于可持续产生烃生物燃料和其它产物的中间体。特别令人感兴趣的是,脱水/脱羧作用形成被称为烃基化物的异戊二烯,其为用于多种合成聚合物(即,合成橡胶)、特殊化学品和汽油添加剂的重要起始原料。虽然异戊二烯可通过分馏石油获得,但是这种材料的纯化是昂贵且耗时的。此外,化石燃料的消耗使得发现这种重要起始原料的另一种来源至关重要。异戊二烯由多种微生物、植物和动物物种天然产生。然而,来自天然存在有机体的异戊二烯的产量在商业上没有吸引力。因此,从生物质衍生的甲羟戊酸内酯大规模合成异戊二烯和其它合成上可用中间体将具有巨大的商业潜力。
技术实现要素:
[0008]
一方面,本发明涉及一种方法,其包含:(a)在固体催化剂存在下,使mvl或包含mvl的溶液反应以得到第一产物或第一产物混合物,其中包含mvl的溶液任选地包含脱水-甲羟戊酸内酯和/或共-反应物。
[0009]
在一些实施例中,共-反应物选自由以下各者组成的群组:水、烷基醇、醚、芳香族化合物、酸、醛、酯及其混合物。在一些实施例中,步骤(a)在约100℃到约500℃范围内的温度下发生。在一些实施例中,步骤(a)在约亚大气压到约200巴范围内的压力下发生。
[0010]
在一些实施例中,固体催化剂为固体酸催化剂。潜在地合适固体催化剂的非限制性说明性列表包括官能化的苯乙烯-二乙烯基苯共聚物、官能化的四氟乙烯-含氟聚合物共
聚物、钙磷灰石、二氧化硅-氧化铝、二氧化硅、二氧化钛、氧化铝、树脂、金属氧化物和/或沸石。
[0011]
在一些实施例中,固体催化剂选自由金属氧化物催化剂和碳催化剂组成的群组。在一些实施例中,固体催化剂的表面积为约20m2/g和约600m2/g之间。在的一些实施例中,固体催化剂选自由以下各者组成的群组:氧化硅、氧化铝、氧化钛、氧化镁、氧化锆、氧化钴、氧化铁、氧化镍、氧化锰、氧化锌、氧化钼、氧化钨、氧化钙、氧化铈、氧化锡和氧化铬及其混合物。在一些实施例中,固体催化剂包含混合型金属氧化物催化剂。
[0012]
在本发明的一些实施例中,固体催化剂包含负载型过渡金属或贵金属,例如,钯、镁、钒、铬、锰、铁、钴、镍、铜、锌、锆、钼、钌、铑、银、锡、钨、钽、铱、铂和金及其混合物。
[0013]
在一些实施例中,第一产物或第一产物混合物包含以下各者中的一种或多种:异戊二烯、3-甲基-2-丁酮、甲基乙烯基酮、脱水-甲羟戊酸内酯、2-戊烯、2-甲基-1-丁烯、2-甲基-1-戊烯、甲烷、氢气或烷基醇。
[0014]
在本发明的一些实施例中,先前描述方法包含额外的步骤:(b)使所述第一产物或第一产物混合物与第一试剂和第二催化剂反应以得到第二产物或第二产物混合物,其中所述第一试剂为水或烯烃或醇。在一些实施例中,第一试剂为烯烃并且所述第二催化剂选自由以下各者组成的群组:钯、铂、钌、钼、钨、铼、锡和铑烯烃复分解催化剂及其混合物。在一些实施例中,第一试剂为醇并且所述第二催化剂选自由以下各者组成的群组:均相和非均相酸和碱、糖、脂肪酶、离子交换树脂、金属氧化物和沸石及其混合物。
[0015]
在一些实施例中,先前描述步骤(b)在约100℃到约500℃范围内的温度下发生。在一些实施例中,这种方法进一步包含步骤:(c)使所述第二产物或第二产物混合物与第二试剂和第三催化剂反应以得到第三产物或第三产物混合物。
[0016]
在另一方面,本发明提供一种方法,其包含:(a)在约100℃到约500℃范围内的温度下,使mvl或包含mvl的溶液反应以得到第一产物或第一产物混合物。
[0017]
另一方面,本发明提供一种方法,其包含:(a)提供表达生物合成mvl路径的微生物有机体;(b)使微生物有机体在包含合适的碳底物的发酵培养基中生长,由此产生生物基mvl;和(c)使所述生物基mvl反应以得到第一产物或第一产物混合物。步骤(c)任选地在固体催化剂存在下和/或在约100℃到约500℃范围内的温度下发生。
[0018]
在一些实施例中,在上述方法中使用的微生物有机体为包含能够实现生物基mvl产生的代谢修饰的非天然存在的微生物有机体。
[0019]
在一些实施例中,代谢修饰包含选自由以下各者组成的群组的至少一种基因集的表达:i.mvae、mvas;ii.mvae、mvas、atob;iii.atob、hmgr、hmgs;和iv.i、ii或iii的功能性同系物。
[0020]
在一些实施例中,代谢修饰包含对至少一个基因,例如acka-pta、adhe和/或1dha的修饰。
[0021]
在一些实施例中,所用的发酵培养基包含可再生原料。
附图说明
[0022]
图1(图1)为在实例11中使用的高压小规模单元的示意图。
具体实施方式
[0023]
甲羟戊酸、甲羟戊酸酯和甲羟戊酸内酯以ph相关的平衡存在。因此,含有“甲羟戊酸酯”的溶液实际上可含有甲羟戊酸、甲羟戊酸酯和/或甲羟戊酸内酯。此外,根据ph,组分可为盐形式。根据所选择的试剂,抗衡离子可为铵、钠、锂、钾、镁、钙、铝或铯的阳离子。为方便起见,将在本文中包含性地使用甲羟戊酸内酯或“mvl”。
[0024]
甲羟戊酸内酯(mvl)和其相关衍生物,如2,3-脱氢甲羟戊酸内酯(4-甲基-5,6-二氢-2h-吡喃-2-酮;也称为脱水甲羟戊酸内酯,aml),代表着可按工业规模制备的可能丰富的原料。通过发酵的生物基产生允许从如糖、甘油、合成气或甲烷的低挥发性可再生原料高产率并且在成本上有竞争力地产生mvl。由生物基mvl和其相关衍生物产生的化学产品可满足对不基于石油或其它不可再生资源的便宜可再生的消费品和工业产品的需求。随后可将生物基甲羟戊酸内酯转化成各种有用的化合物。在本文以及还在2015年11月12日提交的共同未决us s/n 14/939,437中描述了示例性化合物,所述申请以全文引用的方式并入本文中。
[0025]
如本文所用的术语“生物基”意指从生物前体,并且具体来说如生物质的可再生的生物碳源(与不可再生的基于石油的碳源相反)合成化合物。astm设定了用于计算组合物中所包括的生物基材料的水平的方法标准:astm d6866-使用放射性碳分析测定固体、液体和气体样品的生物基含量的标准测试方法。组合物的生物基含量是以材料中的总有机碳的重量(质量)分率或重量(质量)百分比表示的材料中的生物基碳的量。astm d6866类似于不用年龄方程的放射性碳年代测定法。它通过测定材料中的放射性碳(14c)的量与现代参考标准的放射性碳(14c)的量的比率来进行。该比率以百分比形式报告,该百分比称为现代碳百分比(单位“pmc”)。如果所分析的材料为如今的放射性碳和化石碳(即,不含放射性碳)的混合物,那么所获得的pmc值直接与样品中所存在的生物基材料的量相关。
[0026]
astm d6866区分由基于当代基于生物质的输入材料产生的碳与衍生自基于化石的材料的那些碳。生物质含有充分表征量的碳-14,其容易与如不含任何碳-14的化石燃料的其它材料区分。“生物质”一般被定义为用作燃料或能源的植物材料、植被或农业废弃物。对于生物质碳的碳-14同位素与碳-12同位素的比率对于本领域的技术人员来说大体上是已知的,以取自空气样品的碳-14相对于碳-12的当前天然丰度计,为约2
×
10-12
到1。因为生物质中的碳-14的量是已知的,所以可容易地从样品中的总有机碳计算可再生来源的碳的百分比。0%碳-14指示材料中完全缺乏碳-14原子,从而指示基于化石或石油的碳源。类似地,100%碳-14(在大气校正之后)指示现代生物基碳源。
[0027]
在放射性碳年代测定法中使用的现代参考标准为nist(美国国家标准与技术研究所)标准,其中已知的放射性碳含量大致等效于公元1950年。选择公元1950年是因为它代表了在热核武器测试之前的时间,所述热核武器测试随着每次爆炸(称为“碳炸弹”)将大量的过量放射性碳引入到大气中。这是考古学家和地质学家用作参考的时间逻辑点。出于参考,考古学家或地质学家使用放射性碳日期,公元1950年等于“零岁”。它也代表100pmc。在1963年,大气中的“碳炸弹”在测试高峰期和在中断测试的条约之前达到了正常水平的几乎两倍。已粗略估计了自从它出现以来它在大气中的分布,对于从公元1950年活到现在的植物和动物,显示出大于100pmc的值。随着时间推移,它逐渐降低,现在值接近105pmc。这意味着,如玉米、甘蔗或大豆的新鲜生物质材料将提供接近105pmc的放射性碳标记。通过假定约
105pmc代表如今的生物质材料并且0pmc代表石油衍生物,那么针对所述材料所测量的pmc值将反映两个组分类型的比例。举例来说,100%源于如今的大豆的材料将提供接近105pmc的放射性碳标记。但是如果用50%石油碳稀释它,那么它将提供接近53pmc的放射性碳标记。
[0028]
材料的“生物基含量”报告为相关的总可再生有机碳与总有机碳的百分比值。最终结果通过针对材料所测量的pmc值乘以0.95(调整碳炸弹影响)来计算。把最终的%值看作平均生物基结果并且假定在所分析的材料内的所有组分如今还活着(在过去十年内)或者源自化石。一方面,在本发明中使用的材料(例如,前体化合物或所得聚合物)具有大于0%,更优选地大于10%,更优选地大于25%,更优选地大于50%,并且甚至更优选地大于75%的生物基含量。优选地,根据astm d6866,在本发明中使用的材料基本上完全是生物基,这意味着它们具有95%或更高百分比的生物来源。因此,应了解,生物基产物可通过碳指纹识别而与基于石油的产物区分。因此,根据本发明的生物基聚合物和聚合物前体将具有比相同类型的石油(不可再生)来源的聚合物更高的放射性碳-14(14c)含量或更高的14c/12c比率。一方面,生物基聚合物前体和/或所得聚合物将具有大于0,优选地大于1的14c/12c比率。
[0029]
如共同未决us s/n 14/939,437中所述,生物基mvl可充当用于产生若干类别聚合物的常见起始材料。
[0030]
举例来说,mvl和aml自身为用于聚合反应的单体。mvl和aml的内酯结构允许开环共聚合。先前已经报导了mvl衍生的p-甲基-s-戊内酯的开环共聚合。除了开环聚合之外,aml还可为用于自由基聚合反应的共聚单体。在mvl的脱水反应期间,可获得不同的aml异构体,它们通过不饱和位置而不同,即3,4-脱氢甲羟戊酸内酯(4-甲基-3,6-二氢-2h-吡喃-2-酮)、4,5-脱氢甲羟戊酸内酯(4-甲基-3,4-二氢-2h-吡喃-2-酮)以及外-脱氢甲羟戊酸内酯(4-亚甲基四氢-2h-吡喃-2-酮)。这些异构体可用作开环聚合和自由基聚合的单体,与aml类似。
[0031]
除了mvl和上述相关化合物的直接聚合之外,它们还可被转化成用于逐步生成聚合物的构建嵌段,尤其如二醇和二酸。这些转化可按化学反应的类型进行大致分类:i)氧化,ii)还原,iii)内酯开环,iv)烯烃改性,以及v)醇改性。
[0032]
mvl和相关内酯充分氧化以得到α,ω-二羧酸可通过用高价金属氧化物处理来实现,高价金属氧化物尤其如cro6或kmno4,它们还可在化学计量的强氧化剂存在下按催化量采用。用硝酸或分子氧作为氧化剂将伯醇氧化为羧酸也是可能的。可找到用于羧酸以及内酯氧化的以上试剂的实例。仅举几例,mvl衍生的二羧酸可用作用于聚酯、醇酸树脂、不饱和聚酯树脂、聚酯多元醇或聚酰胺的构建嵌段。
[0033]
mvl和相关内酯的充分还原产生被取代的α,ω-二醇。在金属催化剂存在下,如四氢化锂铝的金属氢化物、元素钠或氢气可用于将内酯官能团还原为两个伯醇。这些mvl衍生的二元醇和多元醇可用作尤其聚酯、醇酸树脂、不饱和聚酯树脂、聚酯多元醇、聚碳酸酯或乙烯基-聚氨酯树脂的构建嵌段。
[0034]
如醇、硫醇或胺的亲核试剂能够使内酯开环。mvl和相关内酯与二官能或多官能亲核试剂的反应导致形成被取代的开环的基于mvl的二元醇或多元醇。亲核试剂可为同官能的,尤其例如甘油或乙二胺;或杂官能的,尤其如乙醇胺或巯基乙醇。这些开环衍生物的使
用等效于以上所提到的mvl还原产物。
[0035]
aml和其异构体中的亚烷基的改性尤其包括环氧化、二羟基化、迈克尔加成(michael addition)、狄尔斯-阿尔德尔反应(diels-alder reaction)或[2+2]-环加成。反应产物的官能性多样化,并且可能充当各种聚合物的构建嵌段。内酯官能性在所有以上所提到的反应中都保留。这些化合物的开环聚合可导致形成改性脂肪族聚酯,尤其用作热塑性弹性体、不饱和聚酯树脂或聚氨酯。
[0036]
mvl中的叔醇可经改性形成醚和酯衍生物。可引入促进聚合或如溶解度和粘附性特性的官能团。已报导过,可从mvl获得具有侧接mvl酯基的甲基丙烯酸酯。可设想,这种双官能加成物能够参与自由基和开环聚合,产生丙烯酸树脂和乙烯基聚酯。
[0037]
mvl衍生的二元或多元醇和酸还可充当环氧树脂、聚(乙烯醚)或多官能环碳酸酯的前体。
[0038]
在一些实施例中,本发明提供一种方法,其包含在获得第一产物或第一产物混合物的条件下使mvl或包含mvl的溶液反应的第一步骤。在任选的第二步骤中,任选地在催化剂存在下,第一产物或第一产物混合物可然后进一步与试剂反应。在本发明的一些实施例中,试剂为烯烃或醇。在任选的第三步骤中,由此反应产生的第二产物或第二产物混合物可再次任选地在催化剂存在下与相同或另一种试剂反应以产生第三产物或第三产物混合物。如本文所用,术语“产物混合物”是指结构上不同的化合物的混合物。
[0039]
在本发明的某些方面,如在共同未决us s/n 14/939,437中所描述,产物或产物混合物包含生物基mvl或mvl衍生物的聚合物前体化合物(也称为聚合物构建嵌段)。在一个或多个实施例中,本发明的聚合物前体化合物包含开环的生物基mvl或其衍生物。本发明的聚合物前体化合物的实例包括生物基mvl-二醇、生物基mvl-二酸和生物基mvl-缩水甘油醚/酯。本发明有助于组合物的合成,所述组合物包含开环的生物基mvl化合物中的一种或多种,包括由前述生物基mvl前体化合物中的一种或多种制备的聚合物。本发明进一步提供由如本文所述的生物基mvl或衍生物制备的多种其它生物基聚合物和低聚物。
[0040]
更详细地说,本文描述了衍生自生物基化合物,并且具体来说生物基mvl和其相关衍生物的化合物(例如,单体、低聚物和/或聚合物),和用于合成这些化合物的方法。通过氧化,例如,这些生物基前体可反应以得到用于(不饱和)聚酯、聚酯多元醇和聚酰胺的构建嵌段,以及缩水甘油酯和ω-烯基酯的前体(例如,烯丙醚、高烯丙醚、乙烯醚等)。
[0041]
本发明的方法步骤优选地在溶液中进行,例如在溶剂存在下。在一些实施例中,溶剂通过与mvl、mvl的衍生物或一些衍生自mvl的其它化合物反应而充当共-反应物。在本发明的一些实施例中,水用作溶剂。还可使用其它溶剂,如线性、支化或环状醇或二醇(即,甲醇、乙醇、丙醇、异丙醇、仲丁醇、环丁醇、甘油、2-乙基己醇、丙二醇、丁二醇、十二烷醇等)。一般来说,其它合适的溶剂包括极性非质子溶剂如四氢呋喃(thf)、其它线性或环状醚、二甲亚砜(dmso)、二甲基甲酰胺(dmf)、二噁烷等,烷基醇、醚、芳香族化合物、酸、醛、酯在本发明的各种实施例中都可充当有效的溶剂和共-反应物,如本领域的技术人员可容易地辨别。还可使用水和这些溶剂(和其它合适的溶剂)的混合物。
[0042]
在本发明的一些实施例中,方法步骤中的一个或多个在催化剂存在下发生。催化剂可为当前已知的催化剂或将来开发的催化剂。在一些实施例中,关于第一步骤的优选催化剂,具体地但不排他地为固体催化剂。本发明的优选催化剂包括固体酸催化剂、金属氧化
物催化剂和碳催化剂。固体酸催化剂可包含但不限于一种或多种固体酸材料,无论是现在已知还是将来开发的固体酸材料。可利用的示例性固体酸催化剂包括但不限于杂多酸、酸树脂类型催化剂、内消旋多孔二氧化硅、酸粘土、硫酸化氧化锆、分子筛材料、沸石和在热稳定载体上的酸性材料。在酸性材料提供在热稳定载体上的情况下,热稳定载体可包括例如以下各者中的一种或多种:二氧化硅、氧化锡、氧化铌、氧化锆、氧化钛、碳、α-氧化铝等。可任选地掺杂有额外的酸基团如硫酸盐、磷酸盐等的氧化物自身(例如,zro2、sno2、tio2等)还可用作固体酸催化剂。合适的固体酸催化剂的另外实例包括强酸性离子交换剂如含有磺酸基团的交联的聚苯乙烯。举例来说,amberlyst基团的交联的聚苯乙烯。举例来说,amberlyst为具有不同表面特性和孔隙率的官能化苯乙烯-二乙烯基苯共聚物。官能团一般为硫酸类型的官能团。amberlyst作为蜂窝状或大网状球形珠粒(为密歇根州米德兰市陶氏化学公司(dow chemical co.,midland,mi)的注册商标)。类似地,nation为磺化的基于四氟乙烯的含氟聚合物共聚物,其为固体酸催化剂(为特拉华州威明顿市杜邦公司(e.i.du pont de nemours&co.,wilmington,de)的注册商标)。沸石还可用作固体酸催化剂。其中,h-型沸石一般为优选的,例如丝光沸石组中的沸石或细孔沸石,如沸石x、y和l(即,丝光沸石、毛沸石、菱沸石或八面沸石)。额外的沸石为八面沸石组中已经脱铝的超稳定沸石。固体酸催化剂优选地选自由以下各者组成的群组:官能化苯乙烯-二乙烯基苯共聚物、官能化四氟乙烯-含氟聚合物共聚物、钙磷灰石、二氧化硅-氧化铝、二氧化硅、氧化钛、氧化铝、树脂、金属氧化物和沸石。
[0043]
在本发明的一些优选实施例中,所用的催化剂选自由以下各者组成的群组:氧化硅、氧化铝、氧化钛、氧化镁、氧化锆、氧化钴、氧化铁、氧化镍、氧化锰、氧化锌、氧化钼、氧化钨、氧化钙、氧化铈、氧化锡和氧化铬及其混合物。本发明的一些优选实施例采用包含混合型金属氧化物催化剂的固体催化剂,即,包含两种或更多种不同金属或金属氧化物的催化剂。在一些优选实施例中,本发明方法采用非均相催化剂。在一些实施例中,硫酸氢钾从用于第一催化剂的固体酸催化剂的列表中排除。
[0044]
在一些实施例中,在本发明中使用的固体催化剂包含负载型过渡金属或贵金属。由本发明涵盖的过渡金属或贵金属的说明性非限制性列表包括钯、镁、钒、铬、锰、铁、钴、镍、铜、锌、锆、钼、钌、铑、银、锡、钨、钽、铱、铂和金及其混合物。
[0045]
在本发明的一些实施例中,在第二步骤涉及与烯烃反应的情况下,采用选自由以下各者组成的群组的催化剂:钌、钼、钨、铼、锡和铑烯烃复分解催化剂及其混合物。在其它实施例中,在第二步骤涉及与醇反应的情况下,采用选自由以下各者组成的群组的催化剂:均相和非均相酸和碱、糖、脂肪酶、离子交换树脂、金属氧化物和沸石及其混合物。
[0046]
本发明的催化剂由广泛范围的表面积表征。在一些实施例中,所用的固体催化剂的表面积等于或大于1m2/g、5m2/g、10m2/g、20m2/g、50m2/g、100m2/g或200m2/g。在一些实施例中,所用的固体催化剂的表面积等于或小于50m2/g、100m2/g、200m2/g、600m2/g、1000m2/g或2000m2/g。在一些优选实施例中,作为说明性但非限制性实例,固体催化剂的表面积在20m2/g和600m2/g之间。
[0047]
在一些实施例中,第一产物或第一产物混合物包含以下各者中的一种或多种:异戊二烯、3-甲基-2-丁酮、甲基乙烯基酮、脱水-甲羟戊酸内酯、2-戊烯、2-甲基-1-丁烯、2-甲基-1-戊烯、甲烷、氢气或烷基醇。
[0048]
在一些实施例中,第一产物或第一产物混合物包含独立地选自由以下各者组成的群组的化合物:
[0049][0050]
其中r1、r2、r3、r4和r5独立地选自由以下各者组成的群组:h、支化和未支化的c
1-c3烷基以及支化和未支化的c
2-c4烯基。
[0051]
在优选实施例中,第一产物或第一产物混合物包含独立地选自由以下各者组成的群组的化合物:
[0052][0053]
在一个实施例中,任选的第二步骤的烯烃优选地选自由以下各者组成的群组:支化和未支化的c
2-c
20
烯烃、支化和未支化的c
3-c
20
烯酸、支化和未支化的c
3-c
20
烯酸酯,其中烯烃任选被取代。烯烃更优选地选自由以下各者组成的群组:乙烯、丙烯、1-丁烯、2-丁烯、2-甲基-2-丙烯、丙烯酸、棕榈油酸、油酸、亚麻油酸和二十碳四烯酸及其酯。
[0054]
第二步骤可进一步包含第二催化剂。在一个实施例中,第二催化剂优选地为选自由以下各者组成的群组的烯烃复分解催化剂:钯、铂、钌、钼、钨、铼、锡和铑烯烃复分解催化
剂及其混合物。具体催化剂的实例可见于一级文献,专利文献和综述,如grubbs,r.h.等人的《美国化学学会杂志(j.am.chem.soc.)》2011,133,7490-7496;kadyrov,r.等人的《催化主题(top.catal.)》2012,55,538-542;hoveyada,a.h等人《美国化学学会杂志》2009,757(31),10840-10841;grubbs,r.h.等人的《美国化学学会杂志》2012,134(1),693-699;schrodi,y.等人《清洁(clean)》2008,36(9),669-673;艾勒旺斯可再生科学有限公司(elevance renewable sciences inc.)美国专利申请公开第2013/0289327号;和burk,m.j等人的美国专利申请公开第2009/0155866号。优选催化剂在以下示出(grubbs,r.h.等人《美国化学学会杂志》2011,133,7490-7496)。
[0055]
[0056][0057]
在一个实施例中,第二步骤的醇优选地选自由以下各者组成的群组:支化和未支化的c
1-c
34
烷基醇、支化和未支化的c
2-c
20
烷基二醇以及支化和未支化的c
2-c
34
烯醇。醇更优选地选自由以下各者组成的群组:甲醇、乙醇、甘油、2-乙基己醇、丙二醇、丁二醇和十二烷醇。
[0058]
第二步骤可进一步包含第二催化剂。在一个实施例中,第二催化剂优选地选自由以下各者组成的群组:均相和非均相酸和碱、糖、脂肪酶、离子交换树脂、金属氧化物和沸石。第二催化剂更优选地选自由以下各者组成的群组:koh、koch3、naoch3、naoh、h2so4及其混合物。
[0059]
国际纯粹与应用化学联合会(iupac)对本文所述的一些化合物命名如下:甲羟戊酸:3,5-二羟基-3-甲基戊酸;甲羟戊酸内酯:4-羟基-4-甲基四氢-2h-吡喃-2-酮;脱水甲羟
戊酸内酯:4-甲基-5,6-二氢-2h-吡喃-2-酮(第一个异构体)和4-甲基-3,6-二氢-2h-吡喃-2-酮(第二个异构体);脱水甲羟戊酸:(e)-5-羟基-3-甲基戊-2-烯酸(第一个异构体)和(e)-5-羟基-3-甲基戊-3-烯酸(第二个异构体)。
[0060]
优选的是在第一反应之后并且在第二反应之前,除去存在于含有化合物或化合物混合物的溶液中的至少一部分任何水。水分离步骤为任选的,特别是如果在第二步骤使用耐水催化剂或者稀释水是期望的。
[0061]
本发明的反应优选地在约250k到约1000k范围内的温度下进行。在本发明的一些实施例中,反应步骤在等于或大于约25℃、50℃、100℃、150℃、200℃、300℃、400℃或500℃的温度下执行。在本发明的一些实施例中,反应步骤在小于或等于约100℃、150℃、200℃、300℃、400℃、500℃、600℃或750℃的温度下执行。在本发明的某些优选实施例中采用的温度范围的非限制性但说明性实例包括约100℃到500℃、约150℃到500℃、约200℃到500℃、约250℃到500℃、约100℃到400℃、约150℃到400℃、约200℃到400℃和约250℃到500℃。可在存在或不存在催化剂下执行本发明的反应,并且为一般主题,可在适当催化剂存在下采用低温以充分作用,这可通过本领域技术人员容易地确定。
[0062]
本发明的反应也可在亚大气压到极高气压之间变化的压力范围下执行。在本发明的一些实施例中,反应步骤在大于等于约大气压、2巴、5巴、10巴、20巴、35巴、50巴、100巴、200巴、500巴或甚至更高的压力下执行。在本发明的一些实施例中,反应步骤在小于或等于大气压、2巴、5巴、10巴、20巴、35巴、50巴、100巴、200巴、500巴、或1000巴的压力下执行。在本发明的某些优选实施例中采用的压力范围的非限制性但说明性实例包括0到15巴、2巴到100巴、5巴到100巴、10巴到100巴、20巴到100巴、35巴到100巴和50巴到100巴。在一些情况下,可在存在或不存在催化剂下借助于使用相对高的温度和压力的组合来增强本发明的期望输出。举例来说,在本发明的一些实施例中,一个或多个反应在100℃到500℃范围内的温度和20巴到100巴范围内的压力下或在200℃到500℃范围内的温度和在20巴到100巴范围内的压力下发生。
[0063]
在一个实施例中,由第二步骤得到的产物或产物混合物独立地选自由以下各者组成的群组:
[0064]
[0065]
其中r6独立地选自由以下各者组成的群组:支化和支化的c
1-c
34
烷烃、支化和未支化的c
2-c
20
烷基醇以及支化和未支化的c
2-c
34
烯烃,并且更优选地选自由以下各者组成的群组:甲基、乙基、甘油基、2-乙基己基、2-丙醇基、丁醇基以及十二烷基。
[0066]
在另一实施例中,第二步骤产物或产物混合物可在第三温度和第三压力下与试剂和催化剂组合。在优选实施例中,试剂为烯烃并且催化剂优选地为选自由以下各者组成的群组的烯烃复分解催化剂:钯、铂、钌、钼、钨、铼、锡和铑烯烃复分解催化剂及其混合物。具体催化剂的实例可见于一级文献,专利文献和综述,如grubbs,r.h.等人的《美国化学学会杂志(j.am.chem.soc.)》2011,133,7490-7496;kadyrov,r.等人的《催化主题(top.catal.)》2012,55,538-542;hoveyada,a.h等人《美国化学学会杂志》2009,131(31),10840-10841;grubbs,r.h.等人的《美国化学学会杂志》2012,134(1),693-699;schrodi,y.等人《清洁(clean)》2008,36(9),669-673;艾勒旺斯可再生科学有限公司美国专利申请公开第2013/0289327号;和burk,m.j等人的美国专利申请公开第2009/0155866号。优选的催化剂在流程1中示出(grubbs,r.h.等人《美国化学学会杂志》2011,133,7490-7496)。第三步骤的烯烃优选地选自由以下各者组成的群组:支化和未支化的c
2-c
20
烯烃、支化和未支化的c
3-c
20
烯酸、支化和未支化的c
3-c
20
烯酸酯,其中烯烃任选被取代。烯烃更优选地选自由以下各者组成的群组:乙烯、丙烯、1-丁烯、2-丁烯、2-甲基-2-丙烯、丙烯酸、棕榈油酸、油酸、亚麻油酸和二十碳四烯酸及其酯。
[0067]
在另一优选实施例中,试剂为醇,并且催化剂优选地选自由以下各者组成的群组:均相和非均相酸和碱、糖、脂肪酶、离子交换树脂、金属氧化物和沸石。催化剂更优选地选自由以下各者组成的群组:koh、koch3、naoch、naoh、h2so4、hc1、amberliteamberlystcao-ceo2、zsm-5沸石及其混合物。第三步骤的醇优选地选自由以下各者组成的群组:支化和未支化的c
1-c
34
烷基醇、支化和未支化的c
2-c
20
烷基二醇和/或支化和未支化的c
2-c
34
烯醇。醇更优选地选自由以下各者组成的群组:甲醇、乙醇、甘油、2-乙基己醇、丙二醇、丁二醇和/或十二烷醇。
[0068]
在另一实施例中,由第一步骤产生的产物或产物混合物包含具有c8或更小的分子。根据ph,组分可为盐形式。在一些实施例中,根据所选择的试剂,抗衡离子独立地选自铵、钠、锂、钾、镁、钙、铝和/或铯的阳离子。
[0069]
在一些实施例中,产物或产物混合物包含独立地选自由以下各者组成的群组的化合物:
[0070][0071]
其中r1、r2、r3、r4和r5独立地选自由以下各者组成的群组:h、支化和未支化的c
1-c3烷基以及支化和未支化的c
2-c4烯基。
[0072]
一种用于生物产生化学产物如生物基mvl的有用方法为发酵。发酵程序为本领域普通技术人员众所周知的。用于生物合成产生目标化合物如甲羟戊酸酯的一组互补代谢有机体的发酵可用于例如分批发酵、分批进料发酵或连续发酵中。此外,这些发酵方法中的任
一种还可与适用于发酵程序的众所周知的分离方法如分批分离或连续分离偶联。适用于生物产生目标化合物如甲羟戊酸酯的发酵和分离方法的示例性组合包括例如分批发酵和分批分离、分批发酵和连续分离、分批进料发酵和分批分离、分批进料发酵和连续分离、连续发酵和分批分离或连续发酵和连续分离。分批和连续发酵程序的实例在本领域中是众所周知的。
[0073]
甲羟戊酸酯可由天然和非天然存在的微生物两者产生。在一些实施例中,本发明借助于生物合成mvl路径提供能够表达mvl和/或mvl衍生物的经工程改造非天然存在的微生物。本发明进一步提供在产生生物基mvl和mvl衍生物中使用能够表达并且优选地实际上表达生物合成mvl路径的天然和非天然存在的微生物两者的方法。举例来说,在一些实施例中,本发明提供一种方法,其包含以下步骤:(a)提供表达生物合成mvl路径的微生物有机体;和(b)使微生物有机体在包含合适的碳底物的发酵培养基中生长,由此产生生物基mvl。
[0074]
在本发明的一些优选实施例中,本发明提供一种方法,其包含以下步骤:(a)提供表达生物合成mvl路径的微生物有机体;(b)使微生物有机体在包含合适的碳底物的发酵培养基中生长,由此产生生物基mvl;和(c)使所述生物基mvl反应以得到产物或产物混合物。在一些实施例中,反应在催化剂,优选地固体催化剂存在下发生。在一些实施例中,反应在高温和/或高压下发生。在一些实施例中,此第一产物或产物混合物随后在一个或多个后续反应中进一步反应。举例来说,第一产物或产物混合物可任选地在催化剂存在下,并且任选地在的高温和/或高压下,如上所述,与醇或烯烃反应。
[0075]
在一些实施例中,本发明提供通过引入代谢修饰,例如能够实现生物基mvl产生的代谢修饰产生非天然存在的微生物有机体的方法。在一些优选实施例中,代谢修饰包括以下基因集中的至少一种:i.mvae、mvas;ii.mvae、mvas、atob;和/或iii.atob、hmgr、hmgs,或其功能性同系物(或直系同源物)。短语“功能性同系物”是指由于常见世系和序列保留而类似并且在催化、细胞或生物体水平具有相同或类似功能的多核苷酸或多肽序列。此类代谢修饰可能够实现甲羟戊酸酯的稳定产生。本发明进一步提供所得非天然存在的微生物有机体,和在体现本发明的不同方面的各种方法中使用其的方法。如上文所论述,在以上讨论方法的任一方法中产生的甲羟戊酸酯可用于整合方法中,使得甲羟戊酸酯在接下来步骤之前不被分离或在接下来步骤之前不被分开。
[0076]
在一些实施例中,代谢修饰进一步包含至少一个基因的修饰或破坏。在优选实施例中,基因独立地选自由以下各者组成的群组:adhl-adh7、gpd1、gpd2、acka-pta、adhe和ldha。在一个实施例中,非天然存在的微生物有机体选自由以下各者组成的群组:细菌、酵母、藻类和真菌。示例性细菌包括选自由以下各者组成的群组的物种:大肠杆菌(e.coli)、产琥珀酸厌氧螺菌(a.succiniciproducens)、产琥珀酸放线杆菌(a.succinogenes)、产琥珀酸曼氏杆菌(m.succiniciproducens)、埃特里根瘤菌(r.etli)、枯草杆菌(bacillus subtilis)、谷氨酸棒状杆菌(corynebacterium glutamicum)、氧化葡糖杆菌运动发酵单胞菌(gluconobacter oxydans zymomonas mobilis)、雷特氏乳球菌(lactococcus lactis)、胚芽乳杆菌(lactobacillus plantarum)、天蓝色链霉菌(streptomyces coelicolor)、丙酮丁醇梭杆菌(clostridium acetobutylicum)、梭菌属(clostridium sp.)、细长聚球藻(synechococcus elongatus)、荧光假单胞菌(pseudomonas fluorescens)、甲烷八叠球菌属(methanosarcina sp.)、甲基球菌属(methylococcus sp.)和恶臭假单胞菌
(pseudomonas putida)。示例性酵母包括选自由以下各者组成的群组的物种:酿酒酵母(saccharomyces cerevisiae)、粟酒裂殖酵母(schizosaccharomyces pombe)、克鲁维酵母乳酸(kluyveromyces lactis)、克鲁维酵母(kluyveromyces marxianus)、土曲霉(aspergillus terreus)、黑曲霉(aspergillus niger)、根霉菌属(rhizopus oryzae)、少根根霉菌(rhizopus arrhizus)和毕赤酵母属(pichia sp)。在一些实施例中,微生物有机体为甲烷氧化或光合微生物,如甲烷氧化或光合细菌。
[0077]
在一个实施例中,可利用天然存在的扣囊附膜酵母菌(saccharomycopsis fibuligera)ifo 0107(koike等人《发酵与生物工程杂志(j.ferm.bioeng.)》1989,68(1),58-59)产生甲羟戊酸酯。在另一实施例中,在表达用于mva生物合成的基因的重组大肠杆菌中产生甲羟戊酸酯(mva)。hmg-coa合成酶(mvas)和双官能hmg-coa还原酶/乙酰coaa乙酰转移酶(mvae)和乙酰基coaa乙酰转移酶(atob)经克隆以利用内源乙酰基-coa集合体提供用于在大肠杆菌中产生甲羟戊酸酯的途径。类似方法在文献中得到说明(zhang,k.等人《pnas》2014,777(23),8357-8362;hashimoto,s.-i.等人《生物技术快报(biotech.let.)》2004,26,1487-1491;endo,a等人《发酵与生物工程杂志》1989,68(1),58-59)。
[0078]
在另一实施例中,过表达密码子优化的由t7启动子驱动的粪肠球菌(e.faecalis)mvae和mvas基因的大肠杆菌菌株bl21(de3)经克隆以提供产生甲羟戊酸酯的途径。
[0079]
在另一实施例中,过表达密码子优化的粪肠球菌mvae和mvas基因的大肠杆菌菌株酿酒酵母(s.cerevisiae)cen.pk经克隆以提供产生甲羟戊酸酯的途径。
[0080]
在另一实施例中,过表达密码子优化的均由t7启动子驱动的酿酒酵母hmgs和hmgr基因以及大肠杆菌atob基因的大肠杆菌菌株bl21(de3)经工程改造以提供产生甲羟戊酸酯的途径。
[0081]
在另一实施例中,如在文献中所说明可使用梭菌属mt1243利用合成气体或co2/h2共混物产生甲羟戊酸酯(kiriukhin,m.等人《生物过程和生物系统工程(bioprocess biosyst.eng.)》2014,37,245-260)。合成气体或合成气可从多种含碳材料获得,如煤、酸水解的木质纤维素生物质、来自钢厂排放物的回收气体或天然气。
[0082]
在另一实施例中,过表达密码子优化的均由camv35s启动子驱动的粪肠球菌(e.faecalis)mvas和mvae基因以及大肠杆菌atob基因的原始小球藻(chlorella protothecoides)经工程改造以在光合自养、混合营养或异养条件下提供从co2产生甲羟戊酸酯的途径。
[0083]
在另一实施例中,过表达密码子优化的均由trc启动子驱动的粪肠球菌mvas和mvae基因以及大肠杆菌atob基因的荚膜甲基球菌(methylococcus capsulatus)经工程改造以提供从甲烷或甲醇产生甲羟戊酸酯的途径。
[0084]
产生甲羟戊酸酯的微生物有机体的发酵需要碳和能量的来源,如葡萄糖、蔗糖、木糖、阿拉伯糖、半乳糖、甘露糖、果糖、co、h2或co2和光或上述的组合。供应这些能源的示例性材料和/或底物包括生物质、可再生原料、天然气、生物气、煤和原油。生物质被定义为任何植物衍生的有机材料。最佳地,用于能量的生物质由可持续基体获得,如草本和木本能量作物、农产品和饲料作物、农作物废料和残余物、木质废料和残余物、水生植物和其它废弃物(即,一些城市废料)。可用作原料的生物质来源为纤维素生物质、半纤维素生物质或木质纤维素生物质,如农业残余物(即,麦秸、玉米秆、甘蔗渣、麦或棉木屑)、芦苇草、玉米、麦、棉、
木屑或能量作物(即,芒草)。可再生原料被定义为可再生能源,如衍生自活的有机体的材料或其代谢副产物,包括衍生自生物质的材料。除利用农业残余物以外,用作可再生原料的生长的农作物包括玉米、大豆、柳枝稷、麦、亚麻籽、甘蔗、棕榈油和树木(即,白杨)。
[0085]
生物质和可再生原料为多种碳水化合物的特别有用的来源。具体来说,葡萄糖可从如文献所说明的玉米干燥-研磨方法获得(kwiakowski,j.r.等人《工业原料作物与制品(ind.crops prod.)》2006,23,288-296)。对于玉米秆转化为葡萄糖的实例,参见dudgeon,d.等人,用于木质纤维素生物质生物化学转化为乙醇的方法设计和经济学:玉米秆的稀释酸预处理和酶水解(process design and economics for biochemical conversion of lignocellulosic biomass to ethanol:dilute-acid pretreatment and enzymatic hydrolysis of corn stover)(国家可再生能源实验室(national renewable energy laboratory,nrel),科罗拉多州高登公司(golden,co.),2011)。
[0086]
先前讨论的生物质和可再生原料为多种碳水化合物的有用来源,所述碳水化合物可用于生物合成产生的互补代谢有机体的生长培养基。特别有用的碳源包括含有可用于细胞增殖的不同的简单或复杂碳水化合物的培养基或原料。在一个实施例中,营养物和培养基包含至少一种选自由以下各者组成的群组的碳底物:葡萄糖、蔗糖、木糖、阿拉伯糖、半乳糖、甘露糖、果糖、co和co2。在优选实施例中,营养物和培养基为可再生原料。在一个实施例中,可再生原料衍生自生物质。在优选实施例中,可再生原料选自由纤维素生物质或半纤维素生物质组成的群组。在另一实施例中,可再生原料包含选自由碳水化合物、芳香族化合物和木质素组成的群组的碳源。
[0087]
在审阅本文公开内容和以下提供的工作和/或预示实例之后,本发明的各种实施例的额外优点对于本领域的技术人员将显而易见。应了解,除非本文中另有指示,否则本文中所描述的各种实施例未必相互排斥。举例来说,在一个实施例中描述或描绘的特征也可包括于其它实施例中,但不一定包括于其中。因此,本发明涵盖本文中所述的具体实施例的多种组合和/或整合。
[0088]
如本文所用,短语“和/或”,当在两个或更多个项目的列表中使用时,意指可单独地采用列出的项目中的任一个或可采用列出的项目中的两个或更多个的任何组合。举例来说,如果将组合物描述为含有或排除组分a、b和/或c,那么组合物可含有或排除:单独的a;单独的b;单独的c;a与b的组合;a与c的组合;b与c的组合;或a、b和c的组合。
[0089]
本说明书还使用数值范围以定量与本发明的各种实施例相关的某些参数。应理解,当提供数值范围时,此类范围解释为为仅叙述该范围的下限值的权利要求限制以及仅叙述该范围的上限值的权利要求限制提供文字支持。举例来说,所公开的约10到约100的数值范围为叙述“大于约10”(不具有上限范围)的权利要求及叙述“小于约100”(不具有下限范围)的权利要求提供文字支持。此外,每当提供构成上限或下限的数或数值范围,如“等于或大于100”、“小于或等于100”或“10和100之间”时,应理解,此类限制或数值范围解释为为在数字术语之前并入单词“约”的权利要求限制提供文字支持。举例来说,“在10和100之间”应理解为为叙述“在约10和约100之间”的权利要求限制提供文字支持。
[0090]
实例
[0091]
以下实例阐述根据本发明的优选方法。然而,应理解借助于说明方式提供这些实例,且其中任何实例都不应视为对本发明的总体范围的限制。此外,用纯化的甲羟戊酸酯进
行的所有实例也可用未纯化的材料进行。
[0092]
实例1
[0093]
质粒和大肠杆菌菌株构造
[0094]
粪肠球菌v583中编码mvae(乙酰基-coa乙酰转移酶/hmg-coa还原酶,基因库编号aag02438)和mvas(hmg-coa合成酶,基因库编号aag02439)的基因片段从其基因组dna(从atcc获得)扩增。将这些片段在iptg诱导型trc启动子-lac操纵子的控制下插入到载体(具有pbr322起点主链,安比西林标记物laciq,rrnb转录终止序列)中,以获得质粒pse1(seq id no:1)。
[0095]
使用在sambrook-maniatis(green,m.r.;sambrook,j.编《分子克隆:实验指南(molecular cloning:a laboratory manual)》,第四版,2002)中概述的程序,用质粒pse1对xl-1 blue菌株(enda1 gyra96(nal
r
)thi-1 reca1 rela1 lac glnv44 f'[::tn10 proab
+
laci
q
δ(lacz)m15]hsdr17(rκ-mκ
+
))的化学感受态大肠杆菌细胞进行转型,以获得菌株大肠杆菌-se1。
[0096]
实例2
[0097]
甲羟戊酸内酯产生
[0098]
大肠杆菌-se1菌株在1升锥形瓶中在补充有100μg/升安比西林于250ml培养基中的lb培养基中繁殖,在37℃下在定轨振荡器中在220rpm下培育10小时,达到od600为3。将其用作接种物以便在伊孚森(infors)5lt生物反应器中生产。将1.75升生产培养基(含有15g/l葡萄糖、7g/l kh2po4、1g/l nh4cl、5g/l酵母提取物、1g/l柠檬酸、10mg mnso4、2g/l mgso4、200mg/l feso4以及10mg/l硫胺盐酸盐)与生物反应器中的250ml接种物组合。用20%nh4oh将ph维持在7。将温度维持在32℃。以2升/分钟(lpm)鼓入空气并且将搅拌维持在700rpm。接种后10小时,将1ml的1m iptg添加到生物反应器。视需要添加抗泡沫剂。通过以2小时时间间隔将600g/l葡萄糖溶液添加到生物反应器,将葡萄糖浓度维持在约10g/l。在48小时时停止生物反应器运行。通过使用0.45微滤器从培养液中分离细胞,以获得透明培养液。发现在发酵结束时,甲羟戊酸内酯浓度为40g/l。
[0099]
实例3
[0100]
甲羟戊酸内酯纯化
[0101]
通过在旋转式蒸发器中蒸发将来自实例2的澄清培养液浓缩到400ml体积,并且通过添加20%h2so4酸化到ph 2。添加nacl,直到用nacl使溶液饱和。通过使用四次200ml将甲羟戊酸内酯萃取到乙酸乙酯中以获得800ml乙酸乙酯。在旋转式蒸发器中将其浓缩到200ml。将甲羟戊酸内酯从乙酸乙酯反萃取到150ml 10m naoh溶液中。重复酸化和乙基提取步骤并且将所有乙酸乙酯蒸发以获得纯度超过95%的甲羟戊酸内酯。
[0102]
实例4
[0103]
质粒和蓝藻菌株构造
[0104]
粪肠球菌v583中编码mvae(乙酰基-coa乙酰转移酶/hmg-coa还原酶,基因库编号aag02438)和mvas(hmg-coa合成酶,基因库编号aag02439)的基因片段从其基因组dna(从atcc获得)扩增。将这些片段在组成型启动子psbal的控制下插入到载体mcs中以获得质粒pmse1。
[0105]
根据制造商的程序,使用质粒pmsel将构造体插入到蓝藻菌细长聚球藻菌株
pcc7942基因组中(英杰公司(invitrogen),聚球藻属蛋白质表达试剂盒,公开第man0009792号,第16至17页)以获得菌株细长聚球藻-mse1。
[0106]
实例5
[0107]
光合产生甲羟戊酸内酯
[0108]
细长聚球藻-mse1菌株在补充有50mm nahco3和10mg/l硫胺的bg-11培养基中繁殖。细胞在荧光(55μe s-1
m-2
)下在30℃下培育的在1升roux瓶中的600ml培养基中生长,所述荧光通过放置距细胞培养物15cm且充有含有5%co2的空气的八个86-cm 20-w荧光管提供。每天,从细胞培养物中除去细胞培养物的总体积的1/10。然后,将相同体积的含有0.5m nahco3的新鲜培养基添加到细胞培养物中。每天用10n hc1将具有nahco3的细胞培养物的ph调节到7.5。这进行10天。
[0109]
实例6
[0110]
从乙酸酯产生甲羟戊酸内酯
[0111]
大肠杆菌-se1菌株在15ml管中在补充有100μg/升安比西林于5ml培养基中的lb培养基中繁殖,在37℃下在定轨振荡器中在220rpm下培育10小时,达到od600为3。将其用作接种物以便在250ml摇瓶中生产。将50ml生产培养基(含有5g/l乙酸钠、7g/l kh2po4、1g/l nh4cl、5g/l酵母提取物、1g/l柠檬酸、10mg mnso4、2g/l mgso4、200mg/l feso4以及10mg/l硫胺盐酸盐)与摇瓶中的5ml接种物组合。用20%nh4oh将ph维持在7。将温度维持在32℃。接种后10小时,将25μl的1m iptg添加到烧瓶中。通过以6小时时间间隔向摇瓶中添加300g/l乙酸钠溶液将乙酸盐浓度维持在约2g/l。在72小时时停止运行。通过使用0.45微滤器从培养液中分离细胞,以获得透明培养液。发现在发酵结束时,甲羟戊酸内酯浓度为8g/l。
[0112]
实例7
[0113]
从甘油产生甲羟戊酸内酯
[0114]
大肠杆菌-se1菌株在15ml管中在补充有100μg/升安比西林于5ml培养基中的lb培养基中繁殖,在37℃下在定轨振荡器中在220rpm下培育10小时,达到od600为3。将其用作接种物以便在250ml摇瓶中生产。将50ml生产培养基(含有10g/l甘油、7g/l kh2po4、1g/l nh4cl、5g/l酵母提取物、1g/l柠檬酸、10mg mnso4、2g/l mgso4、200mg/l feso4以及10mg/l硫胺盐酸盐)与摇瓶中的5ml接种物组合。用20%nh4oh将ph维持在7。将温度维持在32℃。接种后10小时,将25μl的1m iptg添加到烧瓶中。通过以6小时时间间隔向摇瓶中添加400g/l甘油水溶液将甘油浓度维持在约5g/l。在72小时时停止运行。通过使用0.45微滤器从培养液中分离细胞,以获得透明培养液。发现在发酵结束时,甲羟戊酸内酯浓度为15g/l。
[0115]
实例8
[0116]
从合成气产生甲羟戊酸内酯
[0117]
从合成气(以各种比率的co2、co和h2的混合物)产生甲羟戊酸酯由kiriukhin等人描述(“通过在合成气或co2/h2共混物的连续发酵期间经工程改造的产乙酸菌生物催化剂产生甲羟戊酸酯(mevalonate production by engineered acetogen biocatalyst during continuous fermentation of syngas or co2/h
2 blend)”《生物过程和生物系统工程》2014,37,245-260),并且以其全文并入本文中。
[0118]
实例9
[0119]
质粒和酵母菌株构造
[0120]
启动子和终止子dna序列由加拿大生物基础公司(biobasic,inc,canada)合成。来自粪肠球菌的用于酶类mvae andmvas-(al 10g)的密码子优化的基因序列通过生物基础公司合成。在tdh3启动子和adh1终止子的控制下编码mvae与在tef1启动子和act1终止子的控制下编码mvas-(al 10g)的序列经克隆成载体(其含有2微米起点,ura3标记物和安比西林标记物)以获得质粒pvs19(seq id no:2)。
[0121]
使用在sambrook-maniatis(green,m.r.;sambrook,j编辑《分子克隆:实验指南(molecular cloning:a laboratory manual)》,第四版本,2002)中概述的程序,用质粒pvs19对酵母酿酒酵母cen.pk2-1c(mata;ura3-52;trpl-289;leu2-3,112;his3a 1;mal2-8
c
;suc2)进行转型,以获得染色酿酒酵母-vs19。
[0122]
实例10
[0123]
从蔗糖产生甲羟戊酸内酯
[0124]
酿酒酵母-vs19菌株在15ml管中在5ml的不含尿嘧啶的cm葡萄糖培养液(teknova目录编号c8140)培养基中繁殖,在30℃下在定轨振荡器中在220rpm下培养72小时。通过使用0.45微滤器从培养液中分离细胞,以获得透明培养液。发现在发酵结束时,甲羟戊酸内酯浓度为1.2g/l。
[0125]
实例11
[0126]
初始反应器条件固定床反应器装置
[0127]
在配备有受到高精确性质量流量控制器控制的三个气体管线和经由高精度泵传送液体原料的一个液体进料管线的高压小规模测试单元(图1)中执行反应。所述单元用不锈钢固定床反应器操作、用三区域燃烧炉进行外部加热,同时反应器的出口流经由热交换器冷却并且被导引到容器系统用于液体和气态产物的分离和收集。用插入于催化床中的热电偶监测反应温度。用气相色谱分析液体产物和气态流两者。
[0128]
催化剂和原料
[0129]
用由格雷斯(grace)供应的可商购非晶形sick/akcb催化剂(davicat sial 3113)和由zeolyst公司供应的可商购zsm-5(sick/akos比率23-cbv2314)进行测试。在两种情况下,催化剂以粉末形式供应。如所提供地使用非晶形sick/akcb。zsm-5催化剂在空气中在500℃下煅烧3h以便从铵转换为h
+
形式。然后在使用之前将样品压碎并且筛分成100至180μm的粒径。用10wt%和20wt%的甲羟戊酸内酯水溶液和10wt%脱氢-甲羟戊酸内酯(也称为脱水-甲羟戊酸内酯)执行测试。
[0130]
催化剂装载量
[0131]
玻璃棉塞首先插入反应器中并且在封装之后,使反应器填充有需要的催化剂量。在顶部插入另一个玻璃棉塞,并且然后将反应器连接到所述单元。
[0132]
实验程序和条件
[0133]
在装载有适当量(14.3g)的催化剂的下流不锈钢固定床反应器中执行甲羟戊酸内酯转化测试。使催化剂在300℃下在空气中原位预处理30min。在引入进料之前,在流动惰性气体(n2)下实现期望的反应温度和压力。在达到期望的反应条件之后,使用高精度泵将甲羟戊酸内酯的水溶液(10wt%甲羟戊酸内酯)进料到填充的管状反应器。为了维持压力,将小流量的n2(50cm3/min)与液体进料一起共进料。使用1h-1
或2h-1
的重量时空速度(whsv)在36巴压力下进行反应。基于总的液体进料(溶液)计算whsv。对于每种情形,在~2小时运行
时间之后,进行稳态活性测量。液体收集在阱(~10℃)中,而气态样品收集在气体采样袋中。
[0134]
产物分析
[0135]
在配备有两个检测器(fid和tcd)和以串联旁路配置的三个柱(ms、porapak n和al2o3/kcl)的gc上执行对气态产物的分析。在gc-ms上分析液体。
[0136]
表1-实例11的反应条件
[0137]
条件催化剂t[℃]压力[巴]whsv[h-1
]进料1非晶形sio2/al2o320036 10%甲羟戊酸内酯水溶液2非晶形sio2/al2o330036 10%甲羟戊酸内酯水溶液3非晶形sio2/al2o340036 10%甲羟戊酸内酯水溶液4zsm-520036 10%甲羟戊酸内酯水溶液5zsm-540036 10%甲羟戊酸内酯水溶液
[0138]
借助gc-ms利用非晶形sio2/al2o3对反应的液体产物的分析显示,在所研究温度中的任一温度下产物中不存在进料(即,甲羟戊酸内酯),这指示在所研究条件中实现了甲羟戊酸内酯的完全转化。通过gc-ms的液体产物和气态产物的组成随温度的变化分别呈现在表2和表3中。
[0139]
表2-通过gc-ms在非晶形sio2/al2o3上获得的液体产物的分析
[0140][0141]
液体层中获得的主要产物为脱水-甲羟戊酸内酯。因此,在低温下仅发生脱水。在气态产物中检测到有限量co2以及一些c6烃。
[0142]
在温度增加到300℃时,液体产物分离成两个不同相:形成顶层的油相和类似于乳液且积聚在底部上的水相。将两相分离并且单独地分析。水相主要由3-甲基-2-丁酮、3-甲基-3-丁烯-1-醇和脱水-甲羟戊酸内酯组成,这表明发生了脱水和开环反应两者。油相非常复杂并且由超过150种的以小浓度的化合物组成。表2中示出的分析表示浓度高于1.5%的检测到的组分。在此相中最丰富的化合物为具有苯环的芳香烃。在300℃下在油相和水相两者中检测到少量的异戊二烯。气态产物(表3)显示co2产生增加(较高程度的剧烈裂解反应)和少量的轻质烃。
[0143]
当温度升高到400℃时,液体产物变回到具有乳液状结构的均匀一相产物。在此情
况下,未检测到脱水-甲羟戊酸内酯,并且主要产物为3-甲基-2-丁酮。还产生了在c1到c5范围内的大量轻质烷烃和烯烃,这指示在此类高温(400℃)下,发生了所进料内酯的大量脱羧和裂解。
[0144]
表3-在非晶形sio2/al2o3上获得的气态产物的分析
[0145]
条件123whsv,h-1
111压力,巴363636温度,℃200300400
ꢀꢀꢀꢀ
gc分析vol%
ꢀꢀꢀ
甲烷
ꢀꢀ
0.234乙烷
ꢀꢀ
0.045乙烯
ꢀꢀ
0.043丙烷
ꢀꢀ
0.04丙烯 0.0240.22异丁烷
ꢀꢀ
0.106正丁烷
ꢀꢀ
0.0091-丁烯 0.0090.071异丁烯
ꢀꢀ
0.035顺-2-丁烯0.2810.751.212异戊烷 0.0050.037正戊烷
ꢀꢀ
0.038c
5+
0.010.1421.037c
6+
1.8941.6210.183co24.35410.9169.744co 0.1240.317n295.61388.86591.398总计102.209102.495104.896
[0146]
利用zsm-5的催化结果
[0147]
在非晶形sio2/al2o3的情况下,用zsm-5催化剂在所研究温度中的任一温度的液体产物中并未检测到未转化的甲羟戊酸内酯。这指示,在用zsm-5研究的条件下也实现了甲羟戊酸内酯的完全转化。通过gc-ms在zsm-5上的液体产物和气态产物的组成随温度的变化分别呈现在表4和表5中。
[0148]
在200℃,反应的液体产物为乳液状均相溶液。所获得的主要产物为无水形式的甲羟戊酸内酯和3-甲基-2-丁酮。在气体产物中检测到有限量的co2。相比于非晶形二氧化硅-氧化铝,其中在200℃仅观察到脱水的甲羟戊酸内酯,更多的酸性zsm-5催化剂不仅催化脱水,也催化脱羧。
[0149]
在400℃,液体产物分离成两个不同相:形成顶层的油相和水乳液状相。将两相再次分离并且单独地分析。水相主要由乙酸/丙酸、丙酮和甲苯组成。相比于200℃和非晶形二
氧化硅氧化铝的结果,未观察到3-甲基-2-丁酮。这指示发生大量的脱羧/裂解反应。油相由芳香族化合物如对-二甲苯、甲苯、1,2,3-三甲基-苯和1-乙基-2-甲基-苯组成。这些芳香族物很可能是烯烃低聚合反应的结果,所述烯烃形成为相对于zsm-5的中间体。如表5所示,在气相中检测到co2和少量的轻质烃(烷烃/烯烃)。
[0150]
表4-通过gc-ms在zsm-5上获得的液体产物的分析
[0151]
[0152][0153]
表5-在zsm-5上获得的气态产物的分析
[0154][0155][0156]
实例12
[0157]
温度对非晶形sio2/al2o3的影响
[0158]
按与实例11中描述的程序类似的程序运行实例12中的反应。
[0159]
表6-在实例12和实例13中的反应条件
[0160][0161]
通过gc-ms在275℃下和325℃下(表6的条件1和条件2)液体产物和气态产物的组成分别呈现在表7和表8中。出于比较原因,还呈现先前研究温度(200℃、300℃和400℃)的结果。借助gc-ms对反应的液体产物的分析显示,在研究温度中的任一温度下产物中不存在进料(即,甲羟戊酸内酯),这指示在所研究条件下实现甲羟戊酸内酯的完全转化。
[0162]
表7-在恒定whsv和压力以及不同温度的实验下通过gc-ms在非晶形sio2/al2o3上
获得的液体产物的分析
[0163][0164][0165]
表8-在恒定whsv和压力以及不同温度的实验下在非晶形sio2/al2o3上获得的气态产物的分析
[0166][0167][0168]
在275℃下获得的主要产物为3-甲基-2-丁酮,同时未形成异戊二烯。在325℃,以可测量的量形成异戊二烯以及3-甲基-2-丁酮、2-甲基-2-丙醇和脱氢-甲羟戊酸内酯。由于脱水似乎甚至在275℃下完成,在325℃下出人意料地存在无水形式。然而,这种不一致性可通过以下事实解释:在275℃下实验的进料为脱氢-甲羟戊酸内酯(在200℃下在asa的先前实验获得),而用脱氢-甲羟戊酸内酯和新鲜进料(即,甲羟戊酸内酯)的混合物进行在325℃下的实验。
[0169]
气态产物(表8)并未显示出co2排放随着温度的恒定趋势,这可再次归因于用于研究条件的不同进料。然而,明显的是,轻质烃的浓度随着温度而增加,这指示较高程度的裂解反应。
[0170]
由于就异戊二烯形成而言在325℃下获得最优结果,所以在非晶形sio2/al2o3上甲羟戊酸内酯的脱羧中,选择这一温度用于进一步研究whs v和进料稀释的影响。
[0171]
实例13
[0172]
在非晶形sio2/al2o3上进料浓度和whsv的影响
[0173]
使用与实例11类似的程序。反应条件描述于表6中表9和表10分别呈现在较高whsv下和在进料中的增加的甲羟戊酸内酯浓度的情况下获得的液体产物和气态产物的组成。出
于比较原因,在第一列中还呈现参考条件(whsv=1h-1
,10%水溶液)。
[0174]
然而,我们在产生异戊二烯方面看到明显的增加,这指示其为中间产物,并且因此在较低滞留时间时是有利的。还观察到2-甲基-2-丙醇浓度的增加,同时3-甲基-2-丁酮的形成似乎不受影响。
[0175]
当甲羟戊酸内酯的进料浓度从10wt%增加到20wt%时,检测到重大变化。在较高浓度进料的情况下,记录到异戊二烯形成的数量级增加。在20wt%甲羟戊酸内酯进料的情况下的唯一其它主要产物为3-甲基-2-丁酮。这些结果表明,为了增加异戊二烯的量,应该采用较高的空间速度和较致密的进料。
[0176]
表9-通过gc-ms在325℃下在不同进料组成和whsv的实验下在非晶形sio2/al2o3上获得的液体产物的分析
[0177]
whsv,h-1
121压力,巴363636温度,℃325325325甲羟戊酸内酯水溶液的浓度,%101020gc-ms分析,面积%
ꢀꢀꢀ
甲羟戊酸内酯
ꢀꢀꢀ
脱氢甲羟戊酸内酯17.55
ꢀꢀ
2-甲基-2-丙醇7.114.807 2-甲基-1,3-丁二烯5.428.23854.3072-甲基-3-丁烯-2-醇3.2
ꢀꢀ
3-甲基-2-丁酮44.5146.50733.2323-甲基-3-丁烯-1-醇2.314.982 乙醇2.565.038 2,2-二甲基-丙醛 8.628 2-(2-甲氧基乙氧基)-乙醇3.45
ꢀꢀ
1-甲基-4-(1甲基乙基)-1,3-环己二烯3.45
ꢀꢀ
丙二醇4.83
ꢀꢀ
其它5.7511.80112.462总计100.13100.00100.00
[0178]
表10-在325℃下在不同进料组成和whsv的实验下在非晶形sio2/al2o3上获得的气态产物的分析
[0179][0180][0181]
对用20%甲羟戊酸内酯水溶液的实验进一步分析
[0182]
如上文所论述,在325℃下用20%的甲羟戊酸内酯水溶液在非晶形sio2/al2o3上执行的实验显示,异戊二烯在液体产物中的高百分比作为在gc-ms分析中异戊二烯与总产物面积峰值的面积%。由于这一条件展现就异戊二烯形成而言最有前景的结果,所以我们执行对液体产物样品的进一步分析,以致力于定量异戊二烯的量。第一步骤由用卡尔-费歇尔(karl-fischer)方法(astm d6304)测量液体产物中水的量。发现在液体产物中水为~94wt%。这指示,反应的主要产物为水并且有机物在液体产物样品中的浓度为<6wt%。此外,我们试图使用gc-ms的异戊二烯标准品执行半定量分析以测量所期望产物的实际浓度。这些结果指出,异戊二烯的实际浓度很可能<3wt%,其中其余部分为3-甲基-2-丁酮和其它未识别的组分。
[0183]
实例14
[0184]
异戊二烯的合成
[0185]
根据实例11,在装载有非晶形sio2/al2o3催化剂的下流不锈钢固定床反应器中执行甲羟戊酸内酯转化为异戊二烯。使将催化剂在300℃下在空气中原位预处理30min。在进料之前,在流动惰性气体(n2)下实现期望的反应温度325℃和压力36巴。在达到期望的反应条件之后,使用高精度泵将甲羟戊酸内酯的水溶液(600g甲羟戊酸内酯/l水溶液)进料到填充的管状反应器。为了维持压力,将小流量的n2(50cm3/min)与液体进料一起共进料。使用2h-1
的重量时空速度(whsv)在36巴的压力下进行反应。基于总的液体进料(溶液)计算whsv。在~2小时运行时间之后,进行稳态活性测量。液体收集在阱(~10℃)中,而气态样品收集在气体采样袋中。
[0186]
改变进料浓度和空间速度的额外实验
[0187]
使用上述装置,将14.3gm的催化剂sio2/al2o3(davicat sial 3113,由格雷斯以粉末形式供应并且按原样使用)装载在固定床反应器中并且在300℃下在空气中原位预处理30min。从实例11获得的纯甲羟戊酸内酯的水溶液在如表11中概括的不同条件下泵送通过反应器。在每个实验中的主要产物为异戊二烯,其中3-甲基-2-丁酮作为副产物。
[0188]
表11-改变进料浓度和空间速度的额外实验
[0189]
条件温度(℃)进料浓度(%)空间速度(whsv h-1
)压力(巴)132530%136232550%136332570%136432530%236532550%236632570%236732530%336832550%336932570%3361032510%336
[0190]
实例15
[0191]
经取代芳香族物的合成
[0192]
如实例11中所述,在装载有非晶形zsm-5催化剂的下流不锈钢固定床反应器中执行甲羟戊酸内酯转化为经取代的芳香族物。使催化剂在300℃下在空气中原位预处理30min。在装载之前,zsm-5催化剂在空气中在500℃下煅烧3小时,以将催化剂从铵调节为h
+
形式。在使用之前,将样品压碎并且筛分成100至180μm的粒径。在进料之前,在流动惰性气体(n2)下实现期望的反应温度(250℃、300℃、350℃)和压力(36巴)。在达到期望的反应条件之后,使用高精度泵将甲羟戊酸内酯的水溶液(10%甲羟戊酸内酯水溶液)进料到填充的管状反应器。为了维持压力,将小流量的n2(50cm3/min)与液体进料一起共进料。使用1h-1
的重量时空速度(whsv)在36巴的压力下进行反应。基于总的液体进料(溶液)计算whsv。在~2小时运行时间之后,进行稳态活性测量。液体收集在阱(~10℃)中,而气态样品收集在气体采样袋中。如通过液相和气相色谱分析,产物为芳香族物和3-甲基-2-丁酮的混合物
[0193]
实例16
[0194]
脱水-甲羟戊酸内酯的合成
[0195]
如实例11中所述,在装载有非晶形zsm-5催化剂的下流不锈钢固定床反应器中执行甲羟戊酸内酯转化为脱水-甲羟戊酸内酯。使催化剂在300℃下在空气中原位预处理30min。在装载之前,zsm-5催化剂在空气中在500℃下煅烧3小时,以将催化剂从铵转换为h
+
形式。在使用之前,将样品压碎并且筛分成100至180μm的粒径。在进料之前,在流动惰性气体(n2)下实现期望的反应温度(70℃、100℃、121℃、150℃)和压力(36巴)。在达到期望的反应条件之后,使用高精度泵将甲羟戊酸内酯的水溶液(10%甲羟戊酸内酯水溶液)进料到填充的管状反应器。为了维持压力,将小流量的n2(50cm3/min)与液体进料一起共进料。使用1h-1
的重量时空速度(whsv)在36巴的压力下进行反应。基于总的液体进料(溶液)计算whsv。在~2小时运行时间之后,进行稳态活性测量。液体收集在阱(~10℃)中,而气态样品收集
在气体采样袋中。如通过液相和气相色谱分析,主要产物为脱水-甲羟戊酸内酯。
[0196]
实例17
[0197]
脱水-甲羟戊酸内酯的合成
[0198]
乙酰乙酰基-coa集合体内源性地通过酶乙酰基-coa乙酰转移酶atob由大肠杆菌产生。首先hmg-coa合成酶(mvas或hmgs)和hmg-coa还原酶(mvae或hmgr)经克隆以提供由此集合体产生甲羟戊酸酯的途径。另外为了最大化甲羟戊酸酯通量,蛋白-蛋白基本局部排列探索工具(blastp)用于识别来自各种有机体的mvas和mvae,如粪肠球菌、金黄葡萄球菌(staphylococcusaureus)、干酪乳杆菌(lactobacillus casei)、甲烷球菌属(methanococcus)maripaludis和甲烷球菌属voltae。组合测试用于识别用于甲羟戊酸酯产生的mvas和mvae的最优集合。为了按比例放大甲羟戊酸酯的产生,使带有来自干酪乳酸杆菌的大肠杆菌菌株在1.3l生物反应器中发酵。为了制备脱水-甲羟戊酸内酯,将固体酸催化剂直接添加到发酵培养液并且加热到回流以催化甲羟戊酸酯的脱水。所得脱水-甲羟戊酸内酯通过溶剂萃取使用氯仿而分离。合并的有机相在真空中浓缩以产生粗产物。
[0199]
实例18
[0200]
用对甲苯磺酸合成脱水-甲羟戊酸内酯
[0201]
在1gm的对甲苯磺酸存在下,将10gm甲羟戊酸内酯(在实例2中获得)在甲苯中回流10小时。我们观察到脱水-甲羟戊酸内酯的85%收率。产物用饱和nahco3洗涤并且通过硅胶塞以获得>95%纯度的脱水-甲羟戊酸内酯(1h nmr)。
[0202]
实例19
[0203]
用乙酸钐合成脱水-甲羟戊酸内酯
[0204]
将200mg甲羟戊酸内酯(在实例2中获得)于1ml小瓶中用氩气鼓泡30min,并且在氩气流下添加5mg乙酸钐。将小瓶密封并且在150℃下放置于振荡器中,持续48小时。如通过nmr验证,获得脱水-甲羟戊酸内酯。
[0205]
实例20
[0206]
用氯化铁合成脱水-甲羟戊酸内酯
[0207]
将200mg甲羟戊酸内酯(在实例2中获得)于1ml小瓶中用氩气鼓泡30min,并且在氩气流下添加4mg fecl3。将小瓶密封并且在150℃下放置于振荡器中,持续48小时。如通过nmr验证,获得脱水-甲羟戊酸内酯。
[0208]
实例21
[0209]
用35合成脱水-甲羟戊酸内酯
[0210]
将500mg甲羟戊酸内酯(在实例2中获得)溶解于10ml水中并且均等地分布到10个小瓶中。向每个小瓶添加63mg的湿式35(由陶氏获得)。在密封小瓶之后并且将其加压到65psi,将5个小瓶加热到90℃并且5个小瓶加热到150℃。以不同时间间隔抽取100ul的样品,猝灭并且用水稀释以及经由lc-ms分析。如在表12中突显,脱水-甲羟戊酸内酯为形成的唯一主要产物。
[0211]
表12用35的脱水-甲羟戊酸内酯形成的分析
[0212][0213]
实例22
[0214]
脱水-甲羟戊酸内酯的乙烯醇分解
[0215]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷(500ml),脱氢-甲羟戊酸内酯(0.86mol)和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。在除去溶剂之后,纯化粗产物。
[0216]
实例23
[0217]
5-羟基-3-甲基-2-(e)-戊烯酸的乙烯醇分解
[0218]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷(500ml),5-羟基-3-甲基-2-(e)-戊烯酸(0.86mol)和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。将溶液用氢氧化钠水溶液(300ml至500ml,1m至5m溶液)处理,并且水层用反应溶剂萃取两次。然后将水层酸化到ph 0-2,并且用二氯甲烷或二乙醚(5
×
100ml)萃取。在除去溶剂之后,纯化粗产物。
[0219]
实例24
[0220]
脱水-甲羟戊酸内酯的甲酯
[0221]
将树脂添加到脱水-甲羟戊酸内酯于甲醇中的溶液,并且将浆液加热到回流。在24h之后,使反应冷却到室温,过滤并且在真空中浓缩以提供粗产物,所述粗产物用于进一步反应而不需要纯化。
[0222]
实例25
[0223]
5-羟基-3-甲基-2-(z)-戊烯酸的甲酯的乙烯醇分解
[0224]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷,来自实例24的5-羟基-3-甲基-2-(z)-戊烯酸的甲酯和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。在除去溶剂之后,纯化粗产物。
[0225]
实例26
[0226]
脱水-甲羟戊酸内酯的乙酯
[0227]
将树脂添加到脱水-甲羟戊酸内酯于乙醇中的溶液中,并且将浆液加热到回流。在24h之后,使反应冷却到室温,过滤并且在真空中浓缩以提供粗产物,所述粗产
物用于进一步反应而不需要纯化。
[0228]
实例27
[0229]
5-羟基-3-甲基-2-(z)-戊烯酸的乙酯的乙烯醇分解
[0230]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷,来自实例26的5-羟基-3-甲基-2-(z)-戊烯酸的乙酯和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。在除去溶剂之后,纯化粗产物。
[0231]
实例28
[0232]
脱水-甲羟戊酸内酯的甘油酯
[0233]
将树脂添加到脱水-甲羟戊酸内酯于甘油中的溶液,并且将浆液加热到回流。在24h之后,使反应冷却到室温,过滤并且在真空中浓缩以提供粗产物,所述粗产物用于进一步反应而不需要纯化。
[0234]
实例29
[0235]
5-羟基-3-甲基-2-(z)-戊烯酸的甘油酯的乙烯醇分解
[0236]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷,来自实例28的5-羟基-3-甲基-2-(z)-戊烯酸的甘油酯和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。在除去溶剂之后,纯化粗产物。
[0237]
实例30
[0238]
5-羟基-3-甲基-2-(e)-戊烯酸的甲酯
[0239]
将树脂添加到5-羟基-3-甲基-2-(e)-戊烯酸于甲醇中的溶液,并且将浆液加热到回流。在24h之后,使反应冷却到室温,过滤并且在真空中浓缩以提供粗产物,所述粗产物用于进一步反应而不需要纯化。
[0240]
实例31
[0241]
5-羟基-3-甲基-2-(e)-戊烯酸的甲酯的乙烯醇分解
[0242]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷,来自实例30的5-羟基-3-甲基-2-(e)-戊烯酸的甲酯和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。在除去溶剂之后,纯化粗产物。
[0243]
实例32
[0244]
5-羟基-3-甲基-2-(e)-戊烯酸的乙酯
[0245]
将树脂添加到5-羟基-3-甲基-2-(e)-戊烯酸于乙醇中的溶液,并且将浆液加热到回流。在24h之后,使反应冷却到室温,过滤并且在真空中浓缩以提供粗产物,所述粗产物用于进一步反应而不需要纯化。
[0246]
实例33
[0247]
5-羟基-3-甲基-2-(e)-戊烯酸乙酯的乙烯醇分解
[0248]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷,来自实例32的5-羟基-3-甲基-2-(e)-戊烯酸的乙酯和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。在除去溶剂之后,纯化粗产物。
[0249]
实例34
[0250]
5-羟基-3-甲基-2-(e)-戊烯酸的甘油酯
[0251]
将树脂添加到5-羟基-3-甲基-2-(e)-戊烯酸于甘油中的溶液,并且将浆液加热到回流。在24h之后,使反应冷却到室温,过滤并且在真空中浓缩以提供粗产物,所述粗产物用于进一步反应而不需要纯化。
[0252]
实例35
[0253]
5-羟基-3-甲基-2-(e)-戊烯酸的甘油酯的乙烯醇分解
[0254]
将由厚壁玻璃构成的1l玻璃反应器在氮气或氩气下装入适当的溶剂,如二氯甲烷或二氯乙烷,来自实例34的5-羟基-3-甲基-2-(e)-戊烯酸的甘油酯和格拉布氏钌复分解催化剂(0.01mol%至1.0mol%)。在氮气下搅拌10min至60min之后,用150psi的乙烯气体对容器加压并且在40℃下使反应搅拌超过高达24小时的时段,或者直到方法监测指示反应完成。然后除去并且回收未使用的乙烯,并且将反应器通向大气。在除去溶剂之后,纯化粗产物。
[0255]
实例36
[0256]
用二氧化硅催化剂高度选择性产生异戊二烯和甲基乙烯基酮
[0257]
将小体积固定床反应器装载有1克的davisil等级62硅胶并且加热到250c的温度,其中1.4sccm氮气载气和2.5ul/min流量的于水中的20wt%甲羟戊酸内酯经由同轴注射注入。此外,保持在60c至70c的后置反应器蒸气阱用于捕获水和脱水的甲羟戊酸内酯以防止使用的在线gc-fid或分析的污染。在这些条件下,如使用在线gc-fid分析的异戊二烯的面积分数为91%,其中7%的峰面积被识别为甲基乙烯基酮。
[0258]
基于峰面积分析,相比于异戊二烯的摩尔当量流量的模型流,转化率大于69%,这显示最小的61%总产率(即每摩尔甲羟戊酸内酯0.61摩尔异戊二烯),其中对异戊二烯的选择性为91%。在每种情况下,“总转化率”意指,在45度柱温情况下在4分钟内可通过gc-fid检测的转化为产物的甲羟戊酸内酯的量,并且明确地不包括转化为脱水单独甲羟戊酸内酯。预期几乎100%的甲羟戊酸内酯被消耗,并且如果其并不完全转换为其它分子,那么将转化为脱水的甲羟戊酸内酯。
[0259]
额外实验改变反应器中滞留时间,显示出在低流动速率下选择性和转化率增加。在250c下在于水中的20wt%甲羟戊酸内酯的2.5ul/min流量和14.05sccm的载气流动速率的情况下显示异戊二烯峰面积为84%,其中甲基乙烯基酮的峰面积为14%。相比于模型流的收率表明,对于56%的最小转化率,异戊二烯总收率超过48%。在一定中间滞留时间下在7.01.4sccm的载气但是其它参数保持不变的情况下,异戊二烯选择性为86%,其中甲基乙烯基酮的峰面积为11%,并且通过相同评估方法,对于至少59%的总产率,总转化率最小为
67%。
[0260]
额外实验改变反应器中的温度,显示出在低温下对异戊二烯的选择性增加。在300c下在10ul/min的于水中的20%甲羟戊酸内酯和28.13sccm的氮气情况下,异戊二烯峰面积为72%,其中对于47%的总产率,估计转化率为至少66%。
[0261]
使用约200mg的aerosil 380氧化硅催化剂,额外实验改变甲羟戊酸内酯在水中的浓度。在300c下在5μl/min的于水中的70%甲羟戊酸内酯和14.05sccm的氮气情况下,产物分布显示出64%异戊二烯以及24%甲基乙烯基酮、2%2-甲基-1-丁烯和2%2-甲基-1-丙烯。随着温度增加,甲基乙烯基酮的分数增加而异戊二烯降低。在350c下,异戊二烯的峰面积为36%,甲基乙烯基酮的峰面积为33%,2-甲基-1-丁烯的峰面积为14%,并且2-甲基-1-丙烯的峰面积为6%。在400c下,异戊二烯的峰面积为21%,甲基乙烯基酮的峰面积为39%,2-甲基-1-丁烯的峰面积为33%,并且2-甲基-1-丙烯的峰面积为6%。
[0262]
额外实验除去载气并且将以2.5ul/min的20%甲羟戊酸内酯流到反应器中。在250c下,异戊二烯峰面积为91%,甲基乙烯基酮峰面积为3%以及2-甲基-1-丙烯峰面积为4%。在甚至更低温下,我们看到对异戊二烯的选择性增加。在200c下,异戊二烯峰面积达到96%,其中甲基乙烯基酮在2.5%。在150c下,异戊二烯峰面积达到98.4%,其中甲基乙烯基酮在1%。
[0263]
因此,通过改变反应器温度和滞留时间,我们可产生具有非凡选择性的异戊二烯,或者切换到中间条件以产生异戊二烯、甲基乙烯基酮、1-甲基-2-丁烯和1-甲基-2-丙烯的期望的产物分布。由于我们的分析工具仅定量地监测低沸点化合物,所以这不应被视为对产物分布的限制。当分析反应器产物时发现的其它产物包括但不限于2-丁烯、1,3-戊二烯、2-甲基-1,3-丁二烯、2-丁酮、1,4-戊二烯-3-酮、1-戊烯-3-酮、1-甲基-1,3-环己二烯、1-甲基-1,4-环己二烯、甲苯、1-甲基环己基-2,4-二烯、苯酚、氯苯、二甲苯、4-戊烯-1-基乙酸酯、1-(1,2-二甲基-环戊-2-烯基)-乙酮、4-乙酰基-1-甲基环己烷和脱水的甲羟戊酸内酯。
[0264]
实例37
[0265]
甲羟戊酸内酯在负载于二氧化硅上的钯上的反应
[0266]
使用与实例36中相同的测试反应器,测试大约500mg的siliacat pdo催化剂,负载于二氧化硅上的钯。在325c下在l0ul/min的于水中的20wt%mvl和28.13sccm的氮气载气情况下,异戊二烯峰面积大约为68%,其中甲基乙烯基酮在5%,2-甲基-1-丁烯在4%,2-甲基-1-戊烯在6%以及2-戊烯在15%。
[0267]
额外实验测试不同温度范围和调节方法。举例来说,当催化剂最初保持在275c时,在l0μl/min的于水中的20wt%mvl和28.13sccm的氮气下,增加温度引起对异戊二烯较大的选择性,在375c下以高产率从几乎为零选择性增加到75%选择性。然而,当催化剂最初保持在400c时,随着温度下降到325c对异戊二烯的选择性增加,其中在375c下峰面积为40%,在350c下峰面积为55%,在325c下峰面积为68%。这指示钯更优选地产生其它异构体,其中钯缓慢地被去活化。
[0268]
额外实验测试不同的运行时间。如在以上温度匀变引导测试中指示,较长运行时间一般导致更多的钯失活和于其它产物相比更高的异戊二烯选择性。其它产物包括2-戊烯、2-甲基-1-丙烯、2-甲基-1-丁烯、甲基乙烯基酮、1,3,5-三丁基-苯和其它苯衍生物。
[0269]
额外实验测试不同的滞留时间。在325c下5ul/min的于水中的20wt%mvl和
14.05sccm的氮气情况下,异戊二烯面积%大约为78%,其中对于甲基乙烯基酮为11%,对于2-甲基-1-丁烯为5%以及对于2-甲基-1-丙烯为6%。当通过增加液体mvl溶液的流动速率到10ul/min和28.13sccm的氮气使滞留时间减半时,异戊二烯峰面积稍微增加到82%,其中对于甲基乙烯基酮为9%,对于2-甲基-1-丁烯为3%以及对于2-甲基-1-丙烯为6%。当使用15ul/min的mvl溶液的流动速率和41.72sccm的氮气使滞留时间减小到1/3时,异戊二烯峰面积%稍微降低到81%,其中甲基乙烯基酮在10%,2-甲基-1-丁烯在3%以及2-甲基-1-丙烯在7%。基于此结果,我们估计对于负载于二氧化硅上的钯优选中等滞留时间。在大约1gm/ml粉末振实密度情况下,500mg催化剂的体积大约为500ul。在325c下,1sccm的氮气为约2ml/min的实际流动速率。在325c下,l ul/min的水流量为大约1ml/min的实际流量。因此,在l0ul/min的于水中的mvl溶液和28.13sccm的氮气情况下,我们可估计实际流动速率为66.26ml/min,对于估计的滞留时间为0.45秒。
[0270]
额外实验测试在水中的不同mvl浓度所有测试在325c和40sccm的氮气下完成。最初测试,10ul/min的100%mvl产生80%的峰面积的异戊二烯,对于约70%的平均值,在86分钟之后峰面积降到58%。甲基乙烯基酮存在约16%的峰面积,2-甲基-1-丁烯存在4%以及2-甲基-1-丙烯存在6%。使100%mvl流动速率降低到5ul/min引起约64%的初始异戊二烯峰面积,在294分钟内降到60%。甲基乙烯基酮存在21%的峰面积,2-甲基-1-丁烯存在7%以及2-甲基-1-丙烯存在7%。使用于水中的70wt%mvl,在5ul/min的流动速率情况下,异戊二烯峰面积稍微提高,约70%的峰面积。
[0271]
实例38
[0272]
甲羟戊酸内酯在氧化铝-二氧化硅上的反应
[0273]
在实例36中所描述的设备中执行在氧化铝-二氧化硅催化剂davicat sial 3113上的测试,除了使用实例11中所描述的反应器进行测试以外。在此随后测试中,在实例37情况下看到类似的运行时间行为,其中对异戊二烯的初始选择性几乎为零,但是在许多连续时间之后选择性提高。
[0274]
davicat的最终表现显示出73%的峰面积为异戊二烯,以及8%为甲基异丙基酮。这与其它测试的最大不同之处在于,甲基异丙基酮以显著的量存在,而其它催化剂和没有催化剂测试几乎没有产生甲基异丙基酮且相反有利于甲基乙烯基酮形成。除实例11至实例14中示出的产物列表以外,我们能够识别1-乙基-n-甲基苯、1,3,5-三甲基苯、1-甲基-3-(l-甲基乙基)苯、1,4-二乙基-苯、4-乙基-1,2-二甲基-苯、1,2,4,5-四甲基-苯、1,2,3,4-四甲基-苯、1,2,4,5-四甲基-苯和邻苯二甲酸二乙酯中的若干种异构体。由于对于davicat sial 3113催化剂强烈依赖于运行时间,所以呈现的数字应仅被视为一般估计值。
[0275]
改变反应器温度的额外实验显示出,在300c,30ul/min的于水中的20wt%mvl以及28.13sccm的氮气下,我们看到异戊二烯峰面积开始在50%并且在运行300分钟内增加到83%,其中甲基异丙基酮在相同时间段中从20%降低到4%。在350c,30ul/min的于水中的20wt%mvl以及28.13sccm的氮气下,异戊二烯峰面积为约73%,而甲基异丙基酮为约8%的峰面积。
[0276]
改变压力的额外实验显示出,较高压力(500psi)导致对异戊二烯的选择性降低而有利于苯衍生物。
[0277]
实例39
[0278]
甲羟戊酸内酯在氧化钛上的反应
[0279]
使用实例36中所描述的反应器,在250c到350c范围内的温度下在10ul/min的20wt%mvl和28.13sccm的氮气情况下用500mg催化剂测试aeroxide氧化钛p25。在350c下,异戊二烯呈现63%的峰面积,其中13%为甲基乙烯基酮、13%为2-甲基-1-丁烯以及8%为2-甲基-1-丙烯。在300c下,异戊二烯呈现68%的峰面积以及12%为甲基乙烯基酮、8%为2-甲基-1-丁烯以及11%为2-甲基-1-丙烯。在250c下,异戊二烯呈现42%的峰面积,其中49%为甲基乙烯基酮和6%为2-甲基-1-丁烯。使用与实例36中所述的相同转化率评估方法,使用模型异戊二烯流并且忽略到amvl的转化率,在250c下的转化率为至少15%,其中到异戊二烯的收率为6%。在300c下转化率为44%,其中到异戊二烯的最小收率为30%。在350c下的转化率为55%,其中到异戊二烯的最少收率为35%。
[0280]
实例40
[0281]
在没有催化剂情况下甲羟戊酸内酯的反应
[0282]
在没有催化剂情况下,mvl溶液通过实例36中所描述的反应器反应,其中在300c到450c范围内的温度下大约3ml的反应器内部体积被加热,而没有装载的催化剂。在所有温度下,于水中的20wt%mvl在28.13sccm的氮气载气情况下以10ul/min流动。在300c下,异戊二烯呈现8%的总峰面积,84%为甲基乙烯基酮以及8%为2-甲基-1-丁烯。基于实例36中描述的模型异戊二烯溶液,我们估计11%总转化率和异戊二烯的收率为1%。在350c下,异戊二烯呈现12%的总峰面积,其中71%为甲基乙烯基酮,14%为2-甲基-1-丁烯以及3%为2-甲基-1-丙烯。估计的转化率为30%,其中异戊二烯收率为3%。在400c下,异戊二烯呈现14%的总峰面积,其中54%为甲基乙烯基酮,25%为2-甲基-1-丁烯以及5%为2-甲基-1-丙烯。估计的转化率为44%,其中异戊二烯的收率>6%。在450c下,异戊二烯呈现12%的总峰面积,其中46%为甲基乙烯基酮,32%为2-甲基-1-丁烯以及5%为2-甲基-1-丙烯。估计的转化率为48%,其中异戊二烯的收率为6%。此结果清晰地表明,在没有催化剂情况下,特别地相比于甲基乙烯基酮,异戊二烯大体上是较不利的。
[0283]
实例41
[0284]
甲羟戊酸内酯在负载于氧化铝上的镍金属上的反应
[0285]
使用实例36中所描述的反应器,mvl在来自阿法埃莎(alfa aesar)目录号31276的在二氧化硅氧化铝上的66
±
5%ni催化剂上反应。在300c下,在蒸气阱之后l0ul/min的20wt%mvl和28.13sccm的氮气在在线fid上仅产生一个可见峰,推测为甲烷、一氧化碳或氢气。在产物流中未识别出异戊二烯、甲基乙烯基酮、甲基异丙基酮或其它已知产物。使反应器温度降低到250c并未对可见产物产生显著变化。
[0286]
实例42
[0287]
甲羟戊酸内酯与乙醇在二氧化硅上的反应
[0288]
使用与实例36中相同的测试反应器,装载1.172gm的davisil等级62硅胶并且保持在300c下。在此实例中,通过在注射泵中混合3.638gm的于水中的20wt%mvl与额外的0.402gm无水乙醇,将乙醇作为共-反应物添加,其以10ul/min与2sccm氮气一起注入到反应器中。在使体系平衡30分钟之后,将全部产物分布收集在二氯甲烷或1-辛醇中以避免由于与溶剂重叠而丢失产物峰。在此实例中,绕过蒸气阱和在线gc-fid以允许全部产物收集包括重质和高沸点分子。独立地采集并且分析在二氯甲烷和1-辛醇中产物的质谱。预期一些
溶剂污染物,以及如同所有质谱标识总是有点模糊的情况一样,因此这不应被视为对使用乙醇或任何其它共反应物产生的可能产物的限制。识别化学品的组合列表为
[0289]
·
环丁醇
[0290]
·
4-戊烯-2-醇,3-甲基-[0291]
·
乙醇
[0292]
·
4-戊烯-1-基乙酸酯
[0293]
·
1,3-二氧戊环,2-庚基-4-苯基-[0294]
·
甲基乙烯基酮
[0295]
·
乙酸乙酯
[0296]
·
1,4-戊二烯-3-酮
[0297]
·
4-戊烯酸乙酯
[0298]
·
苯,[(环己氧基)甲基]-[0299]
·
双环[2.2.2]辛-7-烯-2-酮,5-亚甲基-[0300]
·
2,6-辛二烯-1-醇,2,7-二甲基-[0301]
·
对-甲基-1(7)-烯-9-醇
[0302]
·
3-环己基-1-醇,4-甲基-1-(l-甲基乙基)-,乙酸酯
[0303]
·
2,3-环氧蒈烷,(e)-[0304]
·
环己醇,1-甲基-4-(1-甲基乙烯基)-,乙酸酯
[0305]
·
乙醇,2-(3,3-二甲基环己二烯)-,(z)-[0306]
·
5-蒈醇,反,反-(+)-[0307]
·
2-呋喃酮,2,5-二氢-3,5-二甲基
[0308]
·
3-亚甲基-双环[3.2.1]辛-6-烯-8-醇
[0309]
·
苯乙醇,α,α-二甲基-,乙酸酯
[0310]
·
2-环己-1-酮,4,5-二甲基-[0311]
·
(3s,4r,5r,6r)-4,5-双(羟甲基)-3,6-二甲基环己烯
[0312]
·
4-乙酰基-1-甲基环己烯
[0313]
·
环戊烷,1-乙酰基-1,2-环氧-[0314]
·
酮,1,5-二甲基双环[2.1.0]戊-5-基甲基
[0315]
·
脱氢甲羟戊酸内酯
[0316]
·
螺环[3.4]辛-5-酮
[0317]
·
1-苯并呋喃醇,1,3,3-三甲基-[0318]
·
9-十八烯-12-炔酸,甲酯
[0319]
·
2h-吡喃-2-酮,5,6-二氢-4-(2,3-二甲基-2-丁烯-2-基)-[0320]
·
6-(对甲苯基)-2-甲基-2-庚烯醇
[0321]
·
4,4-二甲基环己二烯酮
[0322]
·
菲并[3,2-b]呋喃-7,11-二酮,1,2,3,4,8,9-六氢-4,4,8-三甲基-,(+)-·
1-壬醇
[0323]
·
癸醛
[0324]
·
1-癸醇
[0325]
·
草酸,异丁基壬基酯
[0326]
·
癸酸,甲酯
[0327]
·
四氢吡喃12-四癸炔-1-醇醚
[0328]
·
碳酸,十八烷基苯基酯
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips