
 商标分类
商标分类  商标转让
商标转让 
取代苯氧酰胺衍生物、应用及用于治疗帕金森病的药物的制作方法
 2021-02-02 02:02:40|
2021-02-02 02:02:40| 290|
290| 起点商标网
起点商标网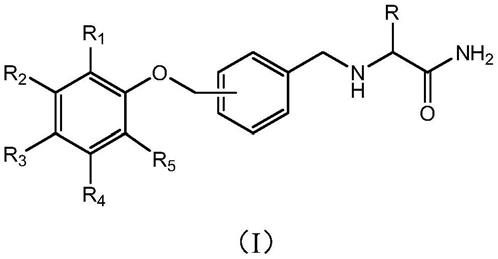
cooh;所述卤素优选氟、氯或溴;
[0013]
所述的给电子基团包括:-nh2,-oh,-och3,-oc2h5,-ch3和-c2h5。
[0014]
可选的,所述吸电子基团选自氟或氯,所述给电子基团选自-och3;
[0015]
优选的,r为甲基。
[0016]
可选的,所述化合物的分子式如ia所示:
[0017][0018]
可选的,所述化合物选自以下结构式所示的化合物:
[0019]
[0020][0021]
可选的,所述化合物的分子式如ib所示:
[0022][0023]
可选的,所述化合物选自以下结构式所示的化合物:
[0024]
[0025][0026]
本发明还提出上述取代苯氧酰胺衍生物在制备用于治疗帕金森病的药物中的应用。
[0027]
本发明还提出一种用于治疗帕金森病的药物组合物,药物组合物包括上述取代苯氧酰胺衍生物;优选的,所述药物组合物中含还有其他活性成分,所述其他活成分为左旋多巴。
[0028]
本发明实施例具有如下有益效果:
[0029]
本发明的取代苯氧酰胺衍生物具有优异的抗mao-b活性,取代苯氧酰胺衍生物还有显著的促细胞增殖作用,对pd神经元细胞的恢复具有积极意义。
附图说明
[0030]
图1为化合物ia-12(200μg/ml)对mpp+(0.1m)诱导pc12细胞凋亡的细胞流式结果;
[0031]
图2为定量分析ia-12(200μg/ml)对mpp+(0.1m)诱导pc12细胞凋亡的影响统计分析图,(n=3,***p<0.001,与生理盐水组比较,&&p<0.01,&p<0.05与mpp+组比较);
[0032]
图3为化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导小鼠帕金森氏病模型的自发活动路线图;
[0033]
图4定量分析化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导小鼠帕金森氏病模型的自发活动影响统计分析图(n=8,***p<0.001,与生理盐水组比较,&&p<0.01,&p<0.05与mptp组比较);
[0034]
图5为化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导小鼠帕金森氏病模型落棒时间的统计分析图,(n=8,***p<0.001,与生理盐水组比较,&&p<0.01,&p<0.05与mptp组比较);
[0035]
图6为化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导pd小鼠纹状体酪氨羟化酶(th)的统计分析图(n=8,***p<0.001,与生理盐水组比较,&&p<0.01,&p<0.05与mptp组比较);
[0036]
图7为化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导pd小鼠纹状体内酪氨羟化酶(th)蛋白表达结果图;
[0037]
图8为化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导纹状体内酪氨羟化酶(th)的统计分析图,(n=8,***p<0.001,与生理盐水组比较,&&p<0.01,&p<0.05与mptp组比较)。
具体实施方式
[0038]
下面结合实施例对本申请进行进一步的介绍。
[0039]
为了更清楚地说明本发明实施例或现有技术中的技术方案,在下述说明中,不同的“一实施例”或“实施例”指的不一定是同一实施例。不同实施例之间可以替换或者合并组合,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些实施例获得其他的实施方式。
[0040]
本发明实施例提出一种取代苯氧酰胺衍生物,分子式为式ⅰ所示的化合物或其药学上可接受的盐:
[0041][0042]
在式ⅰ中:
[0043]
r1选自氢原子、吸电子基团或给电子基团;r2选自氢原子、吸电子基团或给电子基团;r3选自氢原子、吸电子基团或给电子基团;r4选自氢原子、吸电子基团或给电子基团;r5选自氢原子、吸电子基团或给电子基团;
[0044]
r选自氢原子或c
1-c5的直链烷基或支链烷基;与r相连的手性碳的结构为r型或s型。
[0045]
可选的,r1、r2、r3、r4和r5中至少一个选自吸电子基团或给电子基团;优选的,r1、r2、r3、r4和r5中有一个或两个选自吸电子基团或给电子基团。
[0046]
可选的,吸电子基团包括:-no2,-cn,-so3h,-cf3,-ccl3,卤素,-cho和-cooh;卤素优选氟、氯或溴;给电子基团包括:-nh2,-oh,-och3,-oc2h5,-ch3和-c2h5。
[0047]
优选的,吸电子基团选自氟或氯,给电子基团选自-och3;
[0048]
优选的,r为甲基。
[0049]
在本发明实施例中,式ⅰ所示化合物中取代苯氧烷基与中间苯环的取代位置为对位,其结构式如式ia所示:
[0050][0051]
在本发明实施例中,式ⅰ所示化合物中取代苯氧烷基与中间苯环的取代位置为间位,其结构式如ib所示:
[0052][0053]
本发明实施例中的化合物可以是如下结构式所述的化合物:
[0054]
[0055][0056]
本发明实施例还涉及上述取代苯氧酰胺衍生物在制备用于治疗帕金森病的药物中的应用。
[0057]
在本发明的一些实施例中,应用为本发明实施例的取代苯氧酰胺衍生物单独作为活性成分。在本发明的一些实施例中,应用为本发明实施例的取代苯氧酰胺衍生物与活性化合物联合应用。在本发明的一些实施例中,活性化合物为左旋多巴。
[0058]
本发明实施例还涉及一种用于治疗帕金森病的药物,药物的活性组分包括上述取代苯氧酰胺衍生物。
[0059]
下面是上述实施例中ia、ib的制备方法详述:
[0060]
式ia或ib所示的目标化合物的合成以对苯二甲醛(i-1)或间苯二甲醛(i-2)为起始原料,通过还原反应生成对羟甲基苯甲醛(ii-1)或间苯二甲醛(ii-2),ii-1或ii-2与n-溴代丁二酰亚胺(nbs)、三苯基膦(pph3)反应生成对溴甲基苯甲醛(iii-1)或间溴甲基苯甲醛(iii-2),iii-1或iii-2再与取代苯酚、k2co3或cs2co3、ki反应生成对位取代醚中间体(iv-1)或间位取代醚中间体(iv-2),iv-1或iv-2再与α-氨基酰胺盐酸盐、氰基硼氢化钠、三乙胺、型分子筛发生还原氨化反应,得到目标产物ia或ib。合成路线如下所示:
[0061][0062][0063]
在上述合成路线中,r1、r2、r3、r4、r5、r的限定同前所述。
[0064]
本发明实施例中,“药学上可接受的盐”是指保留目标化合物的所需生物活性并表现出最小的不希望的毒理学效应的盐。这些药学上可接受的盐可在该化合物的最终分离和纯化过程中原位制备或者通过单独地将其游离酸或游离碱形式的纯化的化合物分别与合适的碱或酸反应进行制备。
[0065]
本发明实施例化合物的使用剂量和使用方法取决于诸多因素,包括患者的年龄、体重、性别、自然健康状况、营养状况、化合物的活性强度、服用时间、代谢速率、病症的严重
程度以及诊治医师的主观判断。优选的使用剂量介于0.001-1000mg/kg体重/天。该使用量以每天单一剂量进行给药或以每天若干亚剂量进行给药,例如每天给药2、3、4、5或6个剂量。或者所述给药可间歇进行,例如每隔一天一次、每周一次或每月一次。盐或溶剂合物等的治疗有效量可确定为所述化合物本身的治疗有效量的比例。
[0066]
在一些实施方案中,所述药物组合物可任选地还包含一种或多种额外的药物活性化合物。
[0067]
根据本发明,所述药物组合物包含本发明所述化合物以及药学上可接受的载体或赋形剂。该药物组合物可通过例如口服或非肠道等途径给药。本发明的药物组合物可按本领域常规方法制备成各种剂型,包括但不限于片剂、胶囊、溶液、悬浮液、颗粒剂或注射剂等,经例如口服或非肠道等途径给药。
[0068]
本发明所述药物组合物可以每单位剂量含有预定量的活性成分的单位剂型存在。这种单位可含有0.001-1000mg,例如,0.05mg、0.1mg、0.5mg、1mg、10mg、20mg、50mg、80mg、100mg、150mg、200mg、250mg、300mg、500mg、750mg或1g的本发明化合物,其取决于所治疗的疾病、给药途径和受试者的年龄、体重和症状,或者药物组合物可以每单位剂量含有预定量的活性成分的单位剂型存在。在另一实施方案中,所述单位剂量组合物是含有本申请所述的每日剂量或亚剂量或其适当分数的活性成分的那些。此外,这种药物组合物可通过本领域技术人员熟知的任意方法制备。
[0069]
实施例1(2s)-2-(4-(3,4-二氟苯氧甲基)苄基)氨基-丙酰胺(ia-12)的合成
[0070][0071]
1.1 4-羟甲基苯甲醛(ii-1)的合成
[0072]
取一只500ml茄形瓶,将20g对苯二甲醛(0.15mol,4.0equiv)、100ml乙醇和150ml四氢呋喃依次加入瓶中,搅拌溶解均匀。随后在冰浴条件下,一次性缓慢往瓶中加入1.7g硼氢化钠固体(9.3mmol,1.0equiv),反应6h以上,薄层tlc点板,紫外分析仪(254nm)监测反应进程。待对苯二甲醛原料点完全消失后,停止反应,滴加事先配制的2mol/l盐酸溶液进行淬灭,调节ph值至4~5,随后将反应液旋蒸至干,所得残渣用水、乙酸乙酯重溶并加入分液漏斗中。水相用等体积乙酸乙酯萃取2至3次,合并乙酸乙酯层,加入饱和氯化钠水溶液洗。随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约30g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为石油醚:乙酸乙酯=3:1,收集所得单醛基还原反应产物,共得到ii-1白色固体17.6g,产率:86.1%。
[0073]
1.2 4-溴甲基苯甲醛(iii-1)的合成
[0074]
取一只500ml茄形瓶,去皮称取10.0g中间体ii-1(0.073mol;1.0equiv),加入150ml二氯甲烷将其溶解,再在搅拌下加入19.6g n-溴代丁二酰亚胺固体。随后冰浴条件下往茄形瓶中分四批加入三苯基膦固体,总计38.5g(0.146mol;2.0equiv),每一批次间隔半小时,待三苯基膦加毕,撤去冰浴,室温下继续反应3h以上,薄层tlc点板,紫外分析仪(254nm)监测反应进程。待ii-1原料点基本消失后,停止反应,将反应液倒入盛有150ml冷水
的烧杯中,再将水相有机相混合液倒入分液漏斗中萃取,水相用150ml二氯甲烷萃取,合并二氯甲烷层,加入饱和氯化钠水溶液洗。随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约15g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为石油醚:乙酸乙酯=99:1,收集所得苄位醇羟基溴代反应产物,共得到iii-1白色固体8.5g,产率:58.0%。
[0075]
1.3 4-(3,4-二氟苯氧甲基)-苯甲醛(iv-3)的制备
[0076]
取一只250ml茄形瓶,称量1.0g中间体iii-1(5.0mmol;1.0equiv)、0.65g 3,4-二氟苯酚(5.0mmol;1.0equiv)、2.1g碳酸钾(15.0mmol;3.0equiv)、0.6g碘化钾(3.0mmol;0.6equiv)、60ml丙酮加入瓶中,搅拌均匀,随后加热至58℃回流反应24h,薄层tlc点板,紫外分析仪(254nm)监测反应进程。随后将反应液过滤并蒸干,所得残渣用2mol/l naoh水溶液、乙酸乙酯重溶并加入分液漏斗中。水相用等体积乙酸乙酯萃取2至3次,合并乙酸乙酯层,加入饱和氯化钠水溶液洗,随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约5g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为石油醚:乙酸乙酯=10:1,收集所得成醚反应产物,共得到0.46g浅黄色固体iv-3,产率:36.5%。1.4(2s)-2-(4-(3,4-二氟苯氧甲基)苄基)氨基-丙酰胺(ia-12)的制备
[0077]
取一只250ml茄形瓶,称量0.68g l-丙氨酰胺盐酸盐(5.4mmol;1.0equiv)、0.26g氰基硼氢化钠(4.3mmol;0.8equiv)、1g型分子筛、0.5ml三乙胺和50ml甲醇加入瓶中,室温搅拌反应20min,随后迅速加入1.34g中间体iv-3(5.4mmol;1.0equiv),加热至40℃继续反应12h,薄层tlc点板,紫外分析仪(254nm)监测反应进程。待反应结束后将溶液过滤,随后将反应液旋蒸至干,所得残渣用水、乙酸乙酯重溶并加入分液漏斗中。水相用等体积乙酸乙酯萃取2至3次,合并乙酸乙酯层,加入饱和氯化钠水溶液洗。随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约4g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为二氯甲烷:甲醇(采用梯度洗脱法,甲醇与二氯甲烷的体积比在50min内由0%逐渐升高至5%),得到的终产物ia-12为白色固体,合计0.49g,产率36.5%。ms(esi)m/z:(m+h
+
)320.1;1h-nmr(400mhz,dmso-d6):9.14(s,2h),7.91(s,1h),6.80-7.63(m,7h),5.08(s,2h),4.02-4.11(m,2h),3.72-3.74(m,1h),1.39(d,3h,j=6.7hz).
[0078]
实施例2(2s)-2-(3-(3,4-二氟苯氧甲基)苄基)氨基-丙酰胺(ib-6)的合成
[0079][0080]
2.1 3-羟甲基苯甲醛(ii-2)的制备
[0081]
取一只500ml茄形瓶,将20g间苯二甲醛(0.15mol;4.0equiv)、100ml乙醇和150ml四氢呋喃依次加入瓶中,搅拌溶解均匀。随后在冰浴条件下,一次性缓慢往瓶中加入1.7g硼氢化钠固体(9.3mmol;1.0equiv),反应6h以上,薄层tlc点板,紫外分析仪(254nm)监测反应进程。待对苯二甲醛原料点完全消失后,停止反应,滴加事先配制的2mol/l盐酸溶液进行淬
灭,调节ph值至4~5,随后将反应液旋蒸至干,所得残渣用水、乙酸乙酯重溶并加入分液漏斗中。水相用等体积乙酸乙酯萃取2至3次,合并乙酸乙酯层,加入饱和氯化钠水溶液洗。随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约30g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为石油醚:乙酸乙酯=3:1,收集所得单醛基还原反应产物,共得到ii-2白色固体16.4g,产率:80.2%。
[0082]
4.2 3-溴甲基苯甲醛(iii-2)的制备
[0083]
取一只500ml茄形瓶,去皮称取10.0g中间体ii-2(0.073mol;1.0equiv),加入150ml二氯甲烷将1b溶解,再在搅拌下加入19.6g n-溴代丁二酰亚胺固体。随后冰浴条件下往茄形瓶中分四批加入三苯基膦固体,总计38.5g(0.146mol;2.0equiv),每一批次间隔半小时,待三苯基膦加毕,撤去冰浴,室温下继续反应3h以上,薄层tlc点板,紫外分析仪(254nm)监测反应进程。待ii-2原料点基本消失后,停止反应,将反应液倒入盛有150ml冷水的烧杯中,再将水相有机相混合液倒入分液漏斗中萃取,水相用150ml二氯甲烷萃取,合并二氯甲烷层,加入饱和氯化钠水溶液洗。随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约15g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为石油醚:乙酸乙酯=99:1,收集所得苄位醇羟基溴代反应产物,共得到iii-2白色固体6.8g,产率:46.8%。
[0084]
2.1 3-(3,4-二氟苯氧甲基)-苯甲醛(iv-4)的制备
[0085]
取一只150ml茄形瓶,称量1.0g中间体iii-2(5.0mmol;1.0equiv)、0.56g 3,4-二氟苯酚(5.0mmol;1.0equiv)、2.5g碳酸铯(7.5mmol;1.5equiv)、0.6g碘化钾(3.0mmol;0.6equiv)、40ml n,n-二甲基甲酰胺加入瓶中,搅拌均匀,随后加热至40℃反应过夜,薄层tlc点板,紫外分析仪(254nm)监测反应进程。随后将反应液过滤并蒸干,所得残渣用2mol/l naoh水溶液、乙酸乙酯重溶并加入分液漏斗中。水相用等体积乙酸乙酯萃取2至3次,合并乙酸乙酯层,加入饱和氯化钠水溶液洗,随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约5g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为石油醚:乙酸乙酯=10:1,收集所得成醚反应产物,共得到0.37g浅黄色固体iv-4,产率:30.0%。
[0086]
2.2(2s)-2-(3-(3,4-二氟苯氧甲基)苄基)氨基-丙酰胺(ib-6)的制备
[0087]
取一只250ml茄形瓶,称量0.68g l-丙氨酰胺盐酸盐(5.4mmol;1.0equiv)、0.26g氰基硼氢化钠(4.3mmol;0.8equiv)、1g型分子筛、0.5ml三乙胺和50ml甲醇加入瓶中,室温搅拌反应20min,随后迅速加入1.24g中间体iv-4(5.4mmol;1.0equiv),加热至40℃继续反应12h,薄层tlc点板,紫外分析仪(254nm)监测反应进程。随后将反应液过滤并蒸干,所得残渣用2mol/l naoh水溶液、乙酸乙酯重溶并加入分液漏斗中。水相用等体积乙酸乙酯萃取2至3次,合并乙酸乙酯层,加入饱和氯化钠水溶液洗,随后,有机相用无水硫酸钠或无水硫酸镁干燥过夜。滤掉干燥剂,称取60-100目规格硅胶粉约4g加入滤液中,旋蒸至干制砂,进行硅胶柱层析分离,选择的洗脱体系为二氯甲烷:甲醇(采用梯度洗脱法,甲醇与二氯甲烷的体积比在50min内由0%逐渐升高至5%),得到的终产物ib-6为白色固体,合计0.49g,产率30.1%。ms(esi)m/z:(m+h
+
)302.1;1h-nmr(400mhz,dmso-d6):7.36-7.39(m,6h),7.12(s,2h),6.77-6.91(m,2h),5.09(s,2h),3.52-3.70(m,2h),2.99-3.01(m,1h),1.13(d,3h,j=6.7hz).
[0088]
实验例1 mao抑制作用及mao-b选择性研究
[0089]
制备大鼠肝单胺氧化酶(mao),依据单胺类物质在单胺氧化酶的作用下被氧化,同时生成副产物过氧化氢,过氧化氢在过氧化物酶的存在下氧化4-氨基安替比林,生成的氧化产物与香草酸反应,生成在490nm下有吸收的红色染料。如果酶被抑制,则生成的红色染料就少,吸光度值就小。通过吸光度值的变化来确定被测物质对酶的抑制活性。
[0090]
ia,ib所示化合物对mao活力抑制作用研究
[0091]
对以上化合物对mao活力抑制作用研究,发现(10-5
m)ia,ib表现出抑制mao活力作用,可使mao活力分别下降约45
±
3.2%和55
±
6.7%左右,与未处理组比较,差异具有显著性。
[0092]
对mao-a/mao-b的选择性作用研究
[0093]
在测定mao-a和mao-b的活性时,先在酶液中分别按比例加入1:100的50um的氯吉灵(mao-a抑制剂)与帕吉灵(mao-b抑制剂)溶液,37℃水浴反应30min后得到mao-b和mao-a,再按前面的方法测定mao-b和mao-a的活性。如表1所示:
[0094]
表1.ia系列和ib系列化合物对mao-b的抑制作用及mao-b/mao-a选择性的比较研究
[0095][0096]
结果发现ia、ib所示化合物表现出相对强的mao-b的选择性。为阳性对照药sal的2.45和2.46倍。
[0097]
实施例2 mptp细胞毒保护作用研究
[0098]
pc12细胞为大鼠肾上腺嗜铬细胞瘤细胞,它能表达酪氨酸羟化酶并且合成多巴胺,因而也被称为多巴胺能细胞。mpp+是1-甲基-4-苯基-1,2,3,6-四氢吡啶(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine,mptp),在体内的代谢产物,以mpp
+
作用于pci2细胞建立的细胞模型已得到国内外学术界公认,并被广泛用于帕金森病的实验研究。利用mpp
+
诱导pc12细胞损伤为pd细胞模型,观察化合物ia-12对mpp
+
的干预作用,探讨新用于治疗pd的可能性。
[0099]
1、采用mpp
+
(100mm)诱导pc12细胞凋亡的模型,结合细胞流式技术,观察zbh-lht-系列对mpp+致细胞损伤及凋亡的保护作用。方法预先半小时加待检测药物ia-12(200μg/ml),然后pc12加入mpp
+
共同孵育24小时,用annexin v-fitc/pi细胞凋亡检测试剂盒(购于roch公司),facscalⅱur流式细胞仪检测细胞的生长活力和凋亡情况,cellquest pro获取数据并进行分析。实验结果如图1和图2所示。
[0100]
mpp
+
(0.1m)浓度作用24h后,如图1的细胞流式检测显示,生理盐水平行对照组凋亡细胞百分数为23.5
±
2.75%,待试药物ia-12(200μg/ml)单独与pc12细胞孵育,凋亡细胞百分数分为28.47
±
3.17%与生理盐水组比较,差异无显著性,提示ia-12(200μg/ml)无显著的细胞毒作用。而mpp
+
模型组细胞凋亡百分比率为49.5
±
10.12%,模型组凋亡细胞显著增加(p<0.001,n=3)。pc12细胞预先与ia-12(200μg/ml)孵育半小时后,再与mpp
+
(0.1m)共同孵育24小时,凋亡细胞百分比降为35.9
±
2.19%,与mpp
+
模型组比较,细胞凋亡显著降
低(p<0.05,n=3)。
[0101]
2、mptp剂量为30mg/kg,腹腔连续注射7天,建立c57/bl小鼠帕金森氏病模型,实验结果如图3和4所示。
[0102]
如图3和图4的自发活动结果显示,生理盐水平行对照组小鼠运动总路程为31.4
±
4.41(m)。mptp组小鼠运动总路程为15.6
±
1.66(m),与生理盐水组相比,差异有显著性(p<0.001,n=8)。给予小鼠待试药物ia-12(20mg/kg)腹腔连续注射7天,小鼠运动总路程为28.8
±
4.23(m),与生理盐水组比较,差异无显著性,提示ia-12(20mg/kg)对小鼠运动能力无影响。在mptp诱导的小鼠帕金森氏病模型上给予待试药物ia-12(20mg/kg),腹腔连续注射7天。小鼠运动总路程为23.9
±
2.66(m),与mptp模型组比较,差异具有显著性(p<0.01,n=8),
[0103]
3、mptp剂量为30mg/kg,腹腔连续注射7天,建立c57/bl小鼠帕金森氏病模型。转棒实验结果见图5所示。
[0104]
如图5的结果显示,生理盐水平行对照组小鼠运动落棒时间为15.3
±
2.11(min)。mptp组小鼠落棒时间为3.5
±
0.86(min),与生理盐水组相比,差异有显著性(p<0.001,n=8)。给予小鼠待试药物ia-12(20mg/kg)腹腔连续注射7天,小鼠落棒时间为13.8
±
5.23(m),与生理盐水组比较,差异无显著性,提示ia-12(20mg/kg)对小鼠运动能力无影响。在mptp诱导的小鼠帕金森氏病模型上给予待试药物ia-12(20mg/kg),腹腔连续注射7天。小鼠落棒时间为11.9
±
2.12(m),与mptp模型组比较,差异具有显著性(p<0.01,n=8)。
[0105]
4、化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导pd小鼠纹状体内酪氨羟化酶(th)蛋白表达结果如图6所示(免疫组化检测),在mptp诱导的pd小鼠纹状体中th阳性细胞显著降低,与对照组相比差异具有统计学意义(p<0.05,n=3)。在mptp诱导的pd小鼠模型上给予待试药物ia-12(20mg/kg),腹腔连续注射7天,小鼠纹状体中th阳性细胞显著增多,与mptp组相比,差异具有统计学意义(p<0.05,n=3)。
[0106]
5、化合物ia-12(20mg/kg)对mptp(30mg/kg)诱导pd小鼠纹状体内酪氨羟化酶(th)蛋白表达情况如图7和图8所示(western blot检测),在mptp诱导的pd小鼠纹状体中th表达处于较低水平,与对照组相比差异具有统计学意义(p<0.05,n=3)。在mptp诱导的pd小鼠模型上给予待试药物ia-12(20mg/kg),腹腔连续注射7天,小鼠纹状体中th表达显著升高,与mptp组相比,差异具有统计学意义(p<0.05,n=3)。
[0107]
实施例3 mptp致c57/bl小鼠帕金森氏病模型药效学评价结果
[0108]
给灵长类和一些啮齿类动物注射1-甲基-4-苯基-1,2,3,6-四氢吡啶(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine,mptp)会不同程度地引起黑质纹状体多巴胺能神经元退行性变化,并产生了在生化、病理和临床特征上酷似pd的表现。mptp损伤模型是pd研究中普遍使用的动物模型,在啮齿类动物中,尤以c57/bl小鼠对mptp最为敏感,mptp模型已成为研究pd发病机制、神经生化、病理解剖、运动及精神障碍、药物作用等方面的最佳工具。
[0109]
在对该动物模型的多种行为学检测方法中,自发活动和转棒法(rotarod)是应用较多的两种学检测方法。本实验采用mptp剂量为30mg/kg,腹腔连续注射7天,建立c57/bl小鼠帕金森氏病模型,
⑴
首先通过动物转棒测试,自发活动等行为学测试,分析对筛选出的ia-12化合物对模型动物行为方面产生的影响。
⑵
行为学实验结束,实验动物断头处死,免疫组化及westblot定量分析纹状体多巴胺能神经元的变化,以确定ia-12对mptp帕金森病
动物药效学产生的作用基础。如表2所示:
[0110]
表2
[0111][0112]
对以上化合物对mao活力抑制作用筛选研究,药物的筛选浓度为10-5
m,发现约10%的筛选分子表现出抑制mao活力作用,可使mao活力下降约30%左右。
[0113]
且有mao-b选择性的候选药物分子有ia-12,ia-13,ia-15可显著抑制mpp+诱导的pc12细胞凋亡,免疫组化及wb实验结果证实ia-12可显著抑制mptp诱导的c57/blc小鼠黑质及纹状体多巴胺能神经元的特异性凋亡,表明ia-12对mptp/mpp+神经毒具有保护作用,行为学研究(自发活动,游泳实验及滚轴试验)结果证实ia-12对可显著改善和纠正mptp/c57/blc帕金森氏病模型的行为学障碍,提升患病小鼠的运动提升能力。提示其对帕金森氏病具有潜在的治疗作用。
[0114]
本发明实施例结果从细胞、离体和在体水平,确定新的取代苯氧酰胺衍生物ia-12对mptp/mpp+神经毒具有保护作用,可显著改善和纠正mptp/c57/blc帕金森氏病模型的行为学障碍,提升患病小鼠的运动提升能力,示其对帕金森氏病具有潜在的治疗作用。
[0115]
应当说明的是,上述实施例均可根据需要自由组合。以上介绍仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips