
 商标分类
商标分类  商标转让
商标转让 
一种含有软相区和硬相区的聚合物粘结剂及其制备方法和应用与流程
 2021-02-02 02:02:40|
2021-02-02 02:02:40| 344|
344| 起点商标网
起点商标网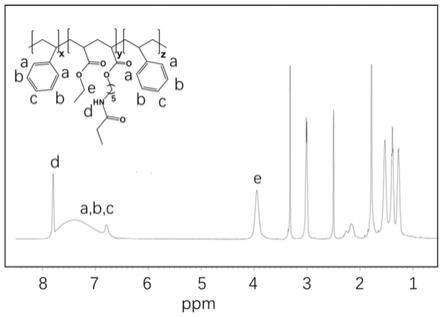
[0001]
本发明属于粘结剂领域,尤其涉及到一种含有软相区和硬相区的聚合物粘结剂及其制备方法和在锂离子电池中的应用。
背景技术:
[0002]
硅基负极的理论容量远大于石墨负极的理论容量,因其具有极高的理论容量而成为替代石墨负极以提高电池能量密度最简单有效的方法。但硅负极的大规模使用目前仍面临巨大的挑战,如硅负极在充放电过程中可以嵌入-脱出更多锂离子,引发巨大的体积膨胀-收缩效应。尤其在嵌锂过程中会产生巨大的体积膨胀,导致硅负极破损,硅负极极片反弹急剧增大甚至引发硅负极活性层从集流体表面脱落的风险。因此,如何有效抑制硅负极体积膨胀,减少sei膜破损成为硅基负极应用发展亟需攻克的技术难题。
[0003]
硅负极粘结剂对于提高负极极片剥离强度,抑制硅负极膨胀,减少硅基负极极片半电/满电反弹具有重要作用。目前传统的cmc、sbr、pvdf等粘结剂已经无法满足硅负极的使用要求,而新型粘结剂如聚丙烯酸、聚丙烯酰胺、聚酰亚胺等刚性偏大,柔韧性不足,在循环过程中仍然存在着因硅负极体积变化粘接失效的风险。因此,需要开发粘结性良好,强度大,能够修复硅负极体积膨胀带来的损伤且柔韧性高的新型粘结剂。
技术实现要素:
[0004]
本发明的目的是提供一种软-硬相区结合的自修复聚合物粘结剂材料,该粘结剂具有高的机械强度、柔韧性和良好的粘结性以及修复硅负极体积膨胀对粘接剂的损伤。其中硬相用来提高粘结剂机械强度并抑制硅负极膨胀,软相用来提高粘结剂的柔韧性和粘结性,并赋予粘结剂自修复性能。
[0005]
本发明目的是通过如下技术方案实现的:
[0006]
一种含有软相区和硬相区的聚合物粘结剂,所述硬相区由刚性单体a聚合得到,所述软相区由柔性单体b和含动态相互作用基团的单体c聚合得到。
[0007]
根据本发明的实施方案,所述粘结剂采用的聚合方式为活性自由基聚合,如可逆加成-断裂链转移聚合(raft)或原子转移自由基聚合(atrp)。
[0008]
根据本发明的实施方案,所述粘结剂的聚合方法,具体步骤如下:
[0009]
先将刚性单体a进行聚合(形成硬相区),刚性单体a聚合完后,再将柔性单体b和含动态相互作用基团的单体c加入继续进行聚合(形成软相区),最终得到同时含有硬相区(刚性单体a为结构单元)和软相区(刚性单体b和含动态相互作用基团的单体c为结构单元)的粘结剂。其中单体聚合方式可以按照形成硬相-软相的形式进行共聚,即先聚合刚性单体a,再聚合柔性单体b和含动态相互作用基团的单体c,也可以按照形成硬相-软相-硬相或软相-硬相-软相的形式进行聚合,以及根据需要以更多相区组合的形式存在。
[0010]
本发明中,刚性单体指该单体纯的聚合物的tg在室温以上;例如,室温处于玻璃
态。
[0011]
本发明中,柔性单体指该单体纯的聚合物的tg在室温以下;例如,室温处于高弹态,一般侧链比较长。
[0012]
具体地,本发明中所述室温是指温度为25℃。所述单体纯的聚合物是指仅由该单体聚合得到的聚合物。
[0013]
本发明中,动态相互作用基团指含有氢键、主客体相互作用、动态共价键、弱配位键等基团,这些基团可以形成动态相互作用,能够在一定温度下重组。
[0014]
根据本发明的实施方案,所述含动态相互作用基团的单体c为含有能够形成氢键的基团的单体。
[0015]
所述含动态相互作用基团的单体c例如选自中的至少一种;优选为其中,m=1~8;x=0~7;y=1~8。
[0016]
根据本发明的实施方案,所述刚性单体a选自根据本发明的实施方案,所述刚性单体a选自中的至少一种;其中,r1为cl、br、i或f;优选为
[0017]
根据本发明的实施方案,所述柔性单体b选自中的至少一种;优选为其中,n=1~8。
[0018]
根据本发明的实施方案,所述粘结剂的结构可以为硬相-软相、硬相-软相-硬相、软相-硬相-软相,也可以根据需要以更多相区组合的形式存在。
[0019]
根据本发明的实施方案,所述硬相区的聚合物分子量mw为:1000-200万,软相区的聚合物分子量mw为:1000-200万。
[0020]
根据本发明的实施方案,所述软相区为由单体b和c聚合形成的无规共聚物单元。
[0021]
根据本发明的实施方案,所述硬相区重复单元占总的重复单元的摩尔比为5-90mol%,如5mol%、10mol%、15mol%、20mol%、25mol%、30mol%、35mol%、40mol%、、45mol%、50mol%、55mol%、60mol%、65mol%、70mol%、75mol%、80mol%、85mol%、90mol%。
[0022]
根据本发明的实施方案,所述软相区重复单元(单体b和单体c聚合形成的重复单元之和)占总的重复单元的摩尔比为10-95mol%,如10mol%、15mol%、20mol%、25mol%、30mol%、35mol%、40mol%、45mol%、50mol%、55mol%、60mol%、65mol%、70mol%、75mol%、80mol%、85mol%、90mol%、95mol%。
[0023]
根据本发明的实施方案,所述单体b和单体c的摩尔比为1-20;示例性为1:1,2:1,3:1,4:1,5:1,6:1,7:1,8:1,10:1,15:1,20:1;优选为7:1。
[0024]
根据本发明的实施方案,所述粘结剂的应力为0.1-20mpa,断裂伸长率为10-200%。
[0025]
本发明的粘结剂中硬相区使粘结剂具有高的机械强度,软相区使粘结剂具有良好的柔韧性,并且软相区存在优异的链段运动能力和一定量的修复基团,使粘结剂具有良好的自修复能力。本发明粘接剂结构示意图如图13所示。
[0026]
本发明还提供上述粘结剂的制备方法,包括将单体a、单体b、单体c进行聚合形成软-硬相区相结合的自修复粘结剂。
[0027]
根据本发明的实施方案,先将单体a进行聚合(形成硬相),单体a聚合完后,再将单体b和c加入继续进行聚合(形成软相),最终得到同时含有硬相(单体a为结构单元)和软相(单体b和c为结构单元)的粘结剂。
[0028]
根据本发明的实施方案,所述聚合方式为活性自由基聚合;例如为可逆加成-断裂链转移聚合(raft)或原子转移自由基聚合(atrp)。
[0029]
根据本发明的实施方案,所述聚合反应的温度为45-100℃;优选为60~90℃;示例性为45℃、60℃、75℃、100℃。
[0030]
根据本发明的实施方案,所述聚合反应的时间为1-24h;优选为4~16h;示例性为1h、2h、4h、6h、8h、10h、12h、16h、20h、24h。
[0031]
根据本发明的实施方案,所述可逆加成-断裂链转移聚合(raft)过程中,需加入可逆加成-断裂链转移试剂(raft试剂)。
[0032]
优选地,所述raft试剂选自三硫代碳酸二苄酯、4-氰基-4-[[(十二烷基硫基)硫代羰基]硫基]戊酸、2-(十二烷基硫基硫代羰基硫基)-2-甲基丙酸、甲基(苯基)氨基二硫代甲酸氰甲酯中的至少一种。
[0033]
优选地,所述raft试剂用量占总的重复单元的摩尔比为0.01%~5%。
[0034]
根据本发明的实施方案,单体a、单体b、单体c聚合过程中需加入引发剂。
[0035]
优选地,所述引发剂为油溶性引发剂;例如,可以为偶氮二异丁腈(aibn)、过氧化苯甲酰、偶氮二异庚腈或偶氮二异氰基戊酸中的至少一种;优选为偶氮二异丁腈(aibn)。
[0036]
根据本发明的实施方案,所述引发剂的用量占总的重复单元的摩尔比为0.01%~
5%。
[0037]
根据本发明的实施方案,所述粘结剂的制备方法还包括将聚合完成后的聚合物溶液进行沉淀、洗涤及真空干燥等步骤。
[0038]
本发明还提供上述粘结剂在锂离子电池电极片中的应用。
[0039]
本发明还提供一种锂离子电池电极片,其含有上述粘结剂;例如,所述锂离子电池电极片可以为正极片和/或负极片;优选地,所述锂离子电池电极片为负极片。
[0040]
根据本发明的实施方案,所述粘结剂在电极片中的质量百分含量为0.1-20wt%。
[0041]
根据本发明的实施方案,所述锂离子电池电极片还包括电极活性物质和/或导电剂。
[0042]
根据本发明的实施方案,所述导电剂为乙炔黑、导电碳球、导电石墨、碳纳米管、导电碳纤维、石墨烯和还原氧化石墨烯中的至少一种。
[0043]
根据本发明的实施方案,所述负极片选自单质硅、氧化亚硅、天然石墨、人造石墨、中间相碳纤维、中间相碳微球、软碳、硬碳中的至少一种。
[0044]
本发明还提供一种锂离子电池电极片的制备方法,其包括将上述粘结剂与电极活性物质和/或导电剂配置成浆料,涂布并烘干;其中,所述粘结剂为浆料总质量的0.1-20wt%。
[0045]
根据本发明的实施方案,含有上述粘结剂的负极片的平均剥离强度为0.1-40n/m。
[0046]
本发明还提供一种锂离子电池,所述锂离子电池含有上述粘结剂和/或上述锂离子电池电极片。
[0047]
根据本发明的实施方案,所述锂离子电池的负极片中含有上述粘结剂。
[0048]
根据本发明的实施方案,所述锂离子电池还包括正极片。
[0049]
本发明的有益效果:
[0050]
本发明所提供的粘结剂中硬相区使粘结剂具有高的机械强度并抑制硅负极膨胀,软相区使粘结剂具有良好的柔韧性和粘结性;并且软相区存在优异的链段运动能力和一定量的修复基团,使粘结剂具有良好的自修复能力,能够及时修复硅负极膨胀造成粘接网络的破损,减少极片的膨胀并延长循环寿命。本发明基于软-硬相结合的自修复粘结剂材料具有高的机械强度、优异的粘结性、良好的柔韧性以及自修复损伤性能。因此,本发明粘结剂可广泛应用于硅负极或者硅/石墨掺混负极,以显著降低负极极片膨胀,并修复硅负极膨胀造成的粘结网络破损,从而显著改善硅负极的循环性能。
附图说明
[0051]
图1为制备例1制得的粘结剂1的核磁谱图(500mhz,dmso-d6);
[0052]
图2为粘结剂1修复前后的应力-应变曲线;
[0053]
图3为制备例2制得的粘结剂2的红外谱图;
[0054]
图4为粘结剂2修复前后的应力-应变曲线;
[0055]
图5为制备例3制得的粘结剂3的核磁谱图(500mhz,cdcl3);
[0056]
图6为粘结剂3修复前后的应力-应变曲线;
[0057]
图7为制备例4制得的粘结剂4的核磁谱图(500mhz,dmso-d6);
[0058]
图8为粘结剂4修复前后的应力-应变曲线;
[0059]
图9为实施例1、对比例1和对比例2的电池放电容量-循环周期曲线;
[0060]
图10为实施例2、对比例1和对比例3的电池放电容量-循环周期曲线;
[0061]
图11为实施例3、对比例1和对比例4的电池放电容量-循环周期曲线;
[0062]
图12为实施例4、对比例1和对比例5的电池放电容量-循环周期曲线;
[0063]
图13为本发明粘接剂结构的示意图。
具体实施方式
[0064]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0065]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0066]
制备例1
[0067]
结构式为的粘结剂1的制备,步骤如下:
[0068]
(1)含硬相区聚合物制备(苯乙烯结构单元作为硬相):称取苯乙烯(10.42g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的聚苯乙烯。
[0069]
(2)硬相-软相区聚合物制备(丙烯酸乙酯结构单元作为软相,5-乙酰胺基丙烯酸戊酯结构单元提供修复基团):称取步骤(1)中制得的聚合物(1.04g),5-乙酰氨基丙烯酸戊酯(1.99g,10mmol),丙烯酸乙酯(7.01g,70mmol),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0070]
(3)含硬相-软相-硬相区聚合物制备:称取步骤(2)中制得的聚合物(10.04g),苯乙烯(1.04g,10mmol),dmf(20ml)和aibn(2.956mg,0.018mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0071]
本实施例获得的粘结剂的核磁谱图如图1所示。
[0072]
修复性能表征:将本实施例获得的粘结剂样品制成标准样品条(长
×
宽
×
厚为
80mm
×
10mm
×
1mm),再用手术刀切成两半,常温下将断面接触不同时间(10min、20min、30min、40min),分别测试不同修复时间后(断面接触不同时间)样品的机械性能,结果如图2所示。从图中结果可以看出,将断面常温修复40min,基本可以达到起始样品的机械性能。由此表明本实施例制得的粘接剂具有快速自修复功能。
[0073]
制备例2
[0074]
结构式为的粘结剂2的制备,步骤如下:
[0075]
(1)含硬相区聚合物制备(丙烯酰胺结构单元作为硬相):称取丙烯酰胺(7.1g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的丙烯酰胺。
[0076]
(2)硬相-软相区聚合物制备(丙烯酸二甲氨基乙酯结构单元作为软相,丙烯酸-2-(3-乙基脲基)乙酯结构单元提供修复基团):称取步骤(1)中制得的聚合物(2.13g),丙烯酸-2-(3-乙基脲基)乙酯(1g,5mmol),丙烯酸二甲氨基乙酯(5.01g,35mmol),dmf(20ml)和aibn(18.06mg,0.11mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0077]
(3)含硬相-软相-硬相区聚合物制备:称取步骤(2)中制得的聚合物(8.14g),丙烯酰胺(2.13g,30mmol),dmf(20ml)和aibn(8.87mg,0.054mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0078]
本实施例获得的粘结剂的核磁谱图如图3所示。
[0079]
修复性能表征:将本实施例获得的粘结剂样品制成标准样品条(长
×
宽
×
厚为80mm
×
10mm
×
1mm),再用手术刀切成两半,常温下将断面接触不同时间(10min、30min),测试修复后样品的机械性能,结果如图4所示。从图中结果可以看出,将断面常温修复30min,其应力可以修复到起始样品的85%,应变可以修复到起始样品应变的82%。
[0080]
制备例3
[0081]
结构式为的粘结剂3的制备:
[0082]
(1)含硬相区聚合物制备(甲基丙烯酸甲酯结构单元作为硬相):称取甲基丙烯酸甲酯(10g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的甲基丙烯酸甲酯。
[0083]
(2)硬相-软相区聚合物制备(丙烯酸丁酯结构单元作为软相,4-酰胺基丙烯酸丁酯结构单元提供修复基团):称取步骤(1)中制得的聚合物(2.0g),4-酰胺基丙烯酸丁酯(1.85g,10mmol),丙烯酸丁酯(6.41g,50mmol),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0084]
(3)含硬相-软相-硬相区聚合物制备:称取步骤(2)中制得的聚合物(10.25g),甲基丙烯酸甲酯(2.0g,20mmol),dmf(20ml)和aibn(2.956mg,0.018mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0085]
本实施例获得的粘结剂的核磁谱图如图5所示。
[0086]
修复性能表征:将本实施例获得的粘结剂样品制成标准样品条(长
×
宽
×
厚为80mm
×
10mm
×
1mm),再用手术刀切成两半,常温下将断面接触1h,测修复后样品的机械性能,结果如图6所示。从图中结果可以看出,将断面常温修复1h,其应力可以修复到起始样品的93%,应变可以修复到起始样品应变的92%。由此表明本实施例制得的粘接剂具有较高的自修复功能。
[0087]
制备例4
[0088]
结构式为的粘结剂4的制备:
[0089]
(1)含硬相区聚合物制备(4-乙烯基吡啶单元作为硬相):称取4-乙烯基吡啶(10.5g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的聚苯乙烯。
[0090]
(2)硬相-软相区聚合物制备(丙烯酸羟乙酯结构单元作为软相,4-乙酰氨基丙烯酸丁酯结构单元提供修复基团):称取步骤(1)中制得的聚合物(2.1g),4-乙酰氨基丙烯酸丁酯(1.86g,10mmol),丙烯酸羟乙酯(5.81g,50mmol),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0091]
(3)含硬相-软相-硬相区聚合物制备:称取步骤(2)中制得的聚合物(9.75g),4-乙烯基吡啶(2.1g,20mmol),dmf(20ml)和aibn(2.956mg,0.018mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0092]
本实施例获得的粘结剂的核磁谱图如图7所示修复性能表征:将样品制成标准样品条(长
×
宽
×
厚为80mm
×
10mm
×
1mm),再用手术刀切成两半,常温下将断面接触不同时间(10min、20min、40min),测修复后样品的机械性能,结果如图8所示。从图中结果可以看出,将断面常温修复40min,其应力可以修复到起始样品的83%,应变可以修复到起始样品应变的87%。
[0093]
制备例5
[0094]
结构式为的粘结剂5(与粘结剂1相比,其不含修复单体5-乙酰胺基丙烯酸戊酯,无修复功能),其制备方法如下:
[0095]
(1)含硬相区聚合物制备(苯乙烯结构单元作为硬相):称取苯乙烯(10.42g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,
0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的聚苯乙烯。
[0096]
(2)硬相-软相区聚合物制备(丙烯酸乙酯结构单元作为软相):称取步骤(1)中制得的聚合物(1.04g),丙烯酸乙酯(8.01g,80mmol),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0097]
(3)含硬相-软相-硬相区聚合物制备:称取步骤(2)中制得的聚合物(10.04g),苯乙烯(1.04g,10mmol),dmf(20ml)和aibn(2.956mg,0.018mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0098]
制备例6
[0099]
结构式为的粘结剂6(与粘结剂2相比,其不含修复单体丙烯酸-2-(3-乙基脲基)乙酯,无修复功能),其制备方法如下:
[0100]
(1)含硬相区聚合物制备(丙烯酰胺结构单元作为硬相):称取丙烯酰胺(7.1g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的丙烯酰胺。
[0101]
(2)硬相-软相区聚合物制备(丙烯酸二甲氨基乙酯结构单元作为软相):称取步骤(1)中制得的聚合物(2.13g),丙烯酸二甲氨基乙酯(5.726g,40mmol),dmf(20ml)和aibn(18.06mg,0.11mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0102]
(3)含硬相-软相-硬相区聚合物制备:称取步骤(2)中制得的聚合物(8.14g),丙烯酰胺(2.13g,30mmol),dmf(20ml)和aibn(8.87mg,0.054mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0103]
制备例7
[0104]
结构式为的粘结剂7,(与粘结剂3相比,不含修复单体4-酰胺基丙烯酸丁酯,无修复功能),其制备方法如下:
[0105]
(1)含硬相区聚合物制备(甲基丙烯酸甲酯结构单元作为硬相):称取甲基丙烯酸甲酯(10g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的甲基丙烯酸甲酯。
[0106]
(2)硬相-软相区聚合物制备(丙烯酸丁酯结构单元作为软相):称取步骤(1)中制得的聚合物(2.0g),丙烯酸丁酯(7.68g,60mmol),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0107]
(3)含硬相-软相-硬相区聚合物制备:称取(2)中制得的聚合物(10.25g),甲基丙烯酸甲酯(2.0g,20mmol),dmf(20ml)和aibn(2.956mg,0.018mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0108]
制备例8
[0109]
结构式为的粘结剂8(与粘结剂4相比,不含修复单体4-乙酰氨基丙烯酸丁酯,无修复功能),其制备方法如下:
[0110]
(1)含硬相区聚合物制备(4-乙烯基吡啶单元作为硬相):称取4-乙烯基吡啶(10.5g,100mmol),三硫代碳酸二苄酯(0.160g,0.55mmol,raft试剂),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥,得到聚合物端基含有raft试剂的聚苯乙烯。
[0111]
(2)硬相-软相区聚合物制备(丙烯酸羟乙酯结构单元作为软相):称取(1)中制得的聚合物(2.1g),丙烯酸羟乙酯(6.96g,60mmol),dmf(20ml)和aibn(29.56mg,0.18mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,室温真空干燥。
[0112]
(3)含硬相-软相-硬相区聚合物制备:称取(2)中制得的聚合物(9.75g),4-乙烯基吡啶(2.1g,20mmol),dmf(20ml)和aibn(2.956mg,0.018mmol)加入到100ml的施兰克瓶中。利用冷冻-解冻法除氧气3次,然后再在75℃下反应12h。聚合反应结束后冷却至室温,将反应溶液加入到去离子水中,使沉淀析出,过滤并用水洗涤多次,80℃真空干燥,得到聚合物粘结剂。
[0113]
实施例1
[0114]
一种锂离子电池,其制备方法步骤如下:
[0115]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例1得到的粘结剂1以质量比为8:1:1分散在n-甲基吡咯烷酮(nmp)溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。
[0116]
采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有氟代碳酸乙烯酯(fec)添加剂(10vol%),将组装好的电池静置12h。
[0117]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图9所示。从图中结果可以看出,电池的首周期放电容量为3510mah/g,循环310周期(t)后放电容量为2120mah/g,容量保持率为60.40%。且极片的起始厚度为41.3μm,循环310t后空电拆解极片厚度为80.3μm,极片反弹率94.43%(表1)。容量保持率和极片反弹率优于对比例1和对比例2。
[0118]
实施例2
[0119]
一种锂离子电池,其制备方法步骤如下:
[0120]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例2得到的粘结剂2以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。
[0121]
采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置12h。
[0122]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图10所示。从图中结果可以看出,电池的首周期放电容量为3515mah/g,循环310周期后放电容量为1986mah/g,容量保持率为50.50%。极片起始厚度为41.1μm,循环310t后空电拆解极片厚度为80.3μm,极片反弹率95.38%(表1)。容量保持率和极片反弹率优于对比例1和对比例3。
[0123]
实施例3
[0124]
一种锂离子电池,其制备方法步骤如下:
[0125]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例3得到的粘结剂3以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。
[0126]
采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置12h。
[0127]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图11所示。从图中结果可以看出,电池的首周期放电容量为3520mah/g,循环310周期后放电容量为1964mah/g,容量保持率为55.80%。极片起始厚度为41.2μm,循环310t后空电拆解极片厚度为81.3μm,极片反弹率97.33%(表1)。容量保持率和极片反弹率优于对比例1和对比例4。
[0128]
实施例4
[0129]
一种锂离子电池,其制备方法步骤如下:
[0130]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例4得到的粘结剂4以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。
[0131]
采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置12h。
[0132]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图12所示。从图中结果可以看出,电池的首周期放电容量为3518mah/g,循环310周期后放电容量为1886mah/g,容量保持率为53.60%。极片起始厚度为41.5μm,循环310t后空电拆解极片厚度为82.3μm,极片反弹率98.31%(表1)。容量保持率和极片反弹率优于对比例1和对比例4。
[0133]
对比例1
[0134]
一种锂离子电池,其制备方法步骤如下:
[0135]
将纳米硅活性物质,导电剂碳黑(super-p)和商品化paa(mw:25w)以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置12h。
[0136]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图9所示。从图中结果可以看出,电池的首周期放电容量为3521mah/g,循环310周期后放电容量为1080mah/g,容量保持率为30.67%。极片起始厚度为41.3μm,循环310t后空电拆解极片厚度为92.3μm,极片反弹率123.49%(表1)。
[0137]
对比例2
[0138]
一种锂离子电池,其制备方法步骤如下:
[0139]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例5得到的粘结剂5以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置
于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置12h。
[0140]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图9所示。从图中结果可以看出,电池的首周期放电容量为3510mah/g,循环310t后放电容量为1490mah/g,容量保持率为42.45%。极片起始厚度为41.4μm,循环310t后空电拆解极片厚度为93.4μm,极片反弹率125.6%(表1)。
[0141]
对比例3
[0142]
一种锂离子电池,其制备方法步骤如下:
[0143]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例6得到的粘结剂6以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置12h。
[0144]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图10所示。从图中结果可以看出,电池的首周期放电容量为3508mah/g,循环310t后放电容量为1010mah/g,容量保持率为28.79%。极片起始厚度为41.6μm,循环310t后空电拆解极片厚度为91.6μm,极片反弹率120.19%(表1)。
[0145]
对比例4
[0146]
一种锂离子电池,其制备方法步骤如下:
[0147]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例7得到的粘结剂7以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置12h。
[0148]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图11所示。从图中结果可以看出,电池的首周期放电容量为3514mah/g,循环310t后放电容量为820mah/g,容量保持率为23.34%。极片起始厚度为41.1μm,循环310t后空电拆解极片厚度为91.2μm,极片反弹率121.90%(表1)。
[0149]
对比例5
[0150]
一种锂离子电池,其制备方法步骤如下:
[0151]
将纳米硅活性物质,导电剂碳黑(super-p)和制备例8得到的粘结剂8以质量比为8:1:1分散在nmp溶剂中,通过研磨和搅拌形成均匀浆料,并涂覆于铜箔上。然后将极片放置于干燥箱中,在80℃下干燥36h,随后裁成直径为1cm的圆形电极片,并置于手套箱中保存(面密度为1mg/cm2)。采用锂片作为对电极,组装2032扣式电池。其中,电解液采用(ec/dmc/dec=1:1:1,1m lipf6)溶液,电解液中含有fec添加剂(10vol%),将组装好的电池静置
12h。
[0152]
将静置好的电池在蓝电测试系统上进行恒流充放电性能测试,充放电电流为500ma/g,电压范围为0.01-1v,测试结果如图12所示。从图中结果可以看出,电池的首周期放电容量为3517mah/g,循环310t后放电容量为1460mah/g,容量保持率为41.51%。极片起始厚度为41.7μm,循环310t后空电拆解极片厚度为92.6μm,极片反弹率122.06%(表1)。
[0153]
表1:实施例1-4,对比例1-5起始极片厚度和循环310周后空电拆解极片厚度和极片膨胀率。
[0154]
实验方案循环前极片厚度310周循环后极片厚度极片反弹率实施例141.3μm80.3μm94.43%实施例241.1μm80.3μm95.38%实施例341.2μm81.3μm97.33%实施例441.5μm82.3μm98.31%对比例141.3μm92.3μm123.49%对比例241.4μm93.4μm125.60%对比例341.6μm91.6μm120.19%对比例441.1μm91.2μm121.90%对比例541.7μm92.6μm122.06%
[0155]
以上,对本发明的实施方式进行了说明。但是,本发明不限定于上述实施方式。凡在本发明的精神和原则之内,所做的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips