
 商标分类
商标分类  商标转让
商标转让 
一种制备2-氯-4-新戊基吡啶的方法与流程
 2021-02-02 01:02:33|
2021-02-02 01:02:33| 384|
384| 起点商标网
起点商标网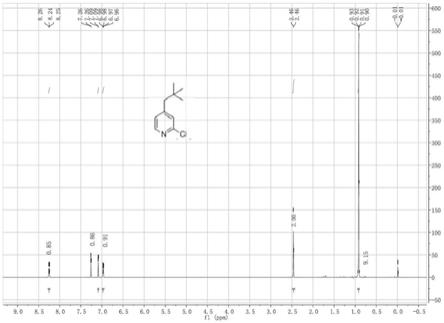
[0001]
本发明属于有机化学合成领域,具体涉及一种制备2-氯-4-新戊基吡啶的方法。
背景技术:
[0002]
有机电致发光器件oled具有效率高亮度、高驱动电压、低响应速度快,以及能实现大面积光电显示等优点,因其在平板显示和高效照明领域具有极大的应用前景而引起广泛关注。在oled的制备及优化中,有机电致发光材料包括小分子和聚合物的选择至关重要。其中有机小分子发光材料具有确定的相对分子质量,化学修饰性强、选择范围广、易于提纯荧光量子产率高以及可以产生红、绿、篮等各种颜色光等优点,作为科研研究的重点方向。论文《有机小分子电致发光材料在oled的发展与应用的综述》中阐述了oled的重要性。
[0003]
目前的类似的有机小分子制备方法有:
[0004]
1)angewandte chemie-international edition,2008,vol.47,43,p.8246-8250中公开了一种以1,6-二氯吡啶和1,1-二甲基丙基氯化镁为原料制备2-氯-6-(叔戊基)吡啶的方法:
[0005][0006]
2)专利wo2012/83224,2012,al公开了用2-溴-6-甲基吡啶和溴代异丁烷为原料制备2-溴-6-异戊基吡啶的方法:
[0007][0008]
目前尚没有制备2-氯-4-新戊基吡啶的方法公开,因此有必要开发一种制备2-氯-4-新戊基吡啶的新方法。
技术实现要素:
[0009]
本发明目的在于提供一种制备2-氯-4-新戊基吡啶的方法,以解决上述背景技术中提出的问题。
[0010]
一种制备2-氯-4-新戊基吡啶的方法,所述方法包括以下步骤:
[0011]
[0012]
步骤(1):将4-吡啶甲醛溶于溶剂中,滴加格式试剂,生成如式(i)所示的4-(2-羟基新戊基)吡啶;
[0013]
步骤(2):将4-(2-羟基新戊基)吡啶与还原剂反应生成如式(ii)所示的化合物4-新戊基吡啶;
[0014]
步骤(3):将4-新戊基吡啶与氧化剂反应生成如式(ⅲ)所示的化合物4-新戊基吡啶氧化物;
[0015]
步骤(4):将4-新戊基吡啶氧化物与氯代试剂反应生成如式(ⅳ)所示的化合物2-氯-4-新戊基吡啶。
[0016]
作为本发明再进一步的方案:步骤(1)中,溶剂可选如下任意一种或多种:四氢呋喃、2-甲基四氢呋喃,甲苯,二甲苯,二氯甲烷、石油醚、乙酸乙酯等;优选四氢呋喃,2-甲基四氢呋喃。
[0017]
作为本发明再进一步的方案:步骤(1)中,滴加格式试剂过程中温度为0~-100℃,优选-40~-30℃。
[0018]
作为本发明再进一步的方案:步骤(1)中,滴加格式试剂结束后,升温至0~100℃,优选40-50℃,反应时间为1~10h,优选2h。
[0019]
作为本发明再进一步的方案:步骤(1)中,所述格式试剂选自叔丁基氯化镁,叔丁基溴化镁等;优选叔丁基氯化镁。
[0020]
作为本发明再进一步的方案:步骤(2)中,还原剂可选氢化铝锂,三乙基硅烷-三氟乙酸混合体系,硼烷,氢碘酸溶液、磷/氢卤酸,氢气/二氧化铂,氢气/钯催化剂等;优选三乙基硅烷-三氟乙酸混合体系。
[0021]
作为本发明再进一步的方案:步骤(2)中,反应温度为50-150℃,优选110-120℃。时间为2~20h,优选4h。
[0022]
作为本发明再进一步的方案:步骤(3)中,氧化剂可选双氧水,高锰酸钾,次氯酸钠,氯酸钾,臭氧,次氯酸,浓硫酸,硝酸,重铬酸钾,2-碘酰基苯甲酸,间氯过氧苯甲酸等;优选双氧水。
[0023]
作为本发明再进一步的方案:步骤(3)中,反应温度为50~150℃,优选70~80℃。时间为10-40h,优选20h。
[0024]
作为本发明再进一步的方案:步骤(3)中反应在有机溶剂中进行;其中有机溶剂可选如下任意一种或多种:四氢呋喃,2-甲基四氢呋喃,甲苯,二甲苯,二氯甲烷,乙酸乙酯,甲醇,乙醇,异丙醇,叔丁醇,n,n-二甲基甲酰胺,二甲基亚砜等,优选甲醇,乙醇。
[0025]
作为本发明再进一步的方案:步骤(4)中,氯代试剂可选氯气,氯化亚砜,三氯氧磷,n-氯代丁二酰亚胺,五氯化磷,三氯化磷,草酰氯,光气磺酰氯等,优选氯化亚砜。
[0026]
作为本发明再进一步的方案:步骤(4)中,反应温度为50~150℃,优选80-90℃。时间为10-40h,优选10h。
[0027]
作为本发明再进一步的方案:步骤(4)中反应在有机介质中进行;有机介质可选如下任意一种或多种:四氢呋喃,2-甲基四氢呋喃,甲苯,二甲苯,氯苯,二氯甲烷,乙酸乙酯,甲醇,乙醇,异丙醇,叔丁醇,n,n-二甲基甲酰胺,二甲基亚砜,氯化亚砜等,优选氯化亚砜。
[0028]
本发明的有益效果为:
[0029]
选用选4-吡啶甲醛作为原料,原料易得,操作简单,产品收率高、纯度高,有利于工
业化生产。
附图说明
[0030]
图1为实例1制备的2-氯-4-新戊基吡啶的核磁谱图;
[0031]
图2为实例1制备的2-氯-4-新戊基吡啶的液相色谱图。
具体实施方式
[0032]
下面将结合实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
[0033]
下述中,如无特殊说明,所有的原料均来自于商购或者通过本领域的常规方法制备而得。
[0034]
所述制备2-氯-4-新戊基吡啶的方法,采用如下的合成路线:
[0035][0036]
实施例1
[0037]
步骤(1):将4-吡啶甲醛(30g)室温溶于四氢呋喃(300ml),体系搅拌溶清,降温并控温-50~-60℃,滴加1.7m叔丁基氯化镁(329.5ml),滴加完毕。自然升温至40-50℃,反应3h。反应结束后降温至0-10℃,滴加饱和氯化铵溶液淬灭反应,搅拌1h,静置分层,水层用乙酸乙酯萃取一次,合并有机层50℃浓缩干,得到44.5g淡黄色固体产物4-(2-羟基新戊基)吡啶,收率96.2%。
[0038]
步骤(2):将上一步得到的产品(40g)和三乙基硅烷(160g)、三氟乙酸(120g)加至反应瓶,升温至(110~120℃)回流反应10h,原料反应完全,降温至50~60℃,加入8ml水,用碳酸钠固体调节至碱性(ph=9~10),再用饱和亚硫酸钠溶液调节体系至淀粉碘化钾试纸不再变色,再加入30ml正己烷,搅拌1h,静置分层,合并有机相,无水硫酸钠干燥。有机层浓缩至干。得到34.1g油状液体产物4-新戊基吡啶,收率94.4%。
[0039]
步骤(3):将上一步得到的产品(30g)和乙醇300ml、双氧水(100ml)加至反应瓶,升温至50~60℃反应20h,反应完全,浓缩乙醇,降至室温,加入二氯甲烷,搅拌,静置分层。有机层浓缩干,得到31.2g油状液体产物4-新戊基吡啶氧化物,收率94.0%。
[0040]
步骤(4):将上一步得到的产品(30g)和氯化亚砜(300ml)加至反应瓶,升温回流反应16h。反应完全,浓缩干。加入150ml的水和150ml二氯甲烷,搅拌0.5h,静置分层。有机层浓缩干,得到30.1g油状液体产物2-氯-4-新戊基吡啶,收率90.3%,液相纯度99.5%。
[0041]1hnmr(400mhz,cdcl3):δppm 8.24(d,j=8.0hz,1h),7.09(d,j=8.0hz,1h),6.97(s,1h),2.46(s,2h),0.91(s,9h)。
[0042]
实施例2
[0043]
步骤(1):将4-吡啶甲醛(40g)室温溶于四氢呋喃(400ml),体系搅拌溶清,降温并控温-30~-40℃,滴加1.7m叔丁基溴化镁(300ml),滴加完毕。自然升温至40-50℃,反应2h。反应结束后降温至0-10℃,滴加饱和氯化铵溶液淬灭反应,搅拌1h,静置分层,水层用乙酸乙酯萃取一次,合并有机层50℃浓缩干,得到53.4g淡黄色固体产物4-(2-羟基新戊基)吡啶,收率90.1%。
[0044]
步骤(2):将上一步得到的产品(50g)和三乙基硅烷(150g)、三氟乙酸(100g)加至反应瓶,升温至(90-100℃)反应20h,原料反应完全,降温至50~60℃,加入8ml水,用碳酸钠固体调节至碱性(ph=9~10),再用饱和亚硫酸钠溶液调节体系至淀粉碘化钾试纸不再变色,再加入100ml正己烷,搅拌1h,静置分层,合并有机相,无水硫酸钠干燥,有机层浓缩至干,得到38.2g油状液体产物4-新戊基吡啶,收率89.5%。
[0045]
步骤(3):将上一步得到的产品(30g)和甲醇300ml、间氯过氧苯甲酸(150g)加至反应瓶,升温至50-60℃反应30h,反应完全,浓缩甲醇,降至室温,加入二氯甲烷,搅拌,静置分层。有机层浓缩干,过柱子得到29.4g油状液体产物4-新戊基吡啶氧化物,收率88.6%。
[0046]
步骤(4):将上一步得到的产品(20g)和三氯氧磷(150ml)加至反应瓶,升温回流反应10h。反应完全,浓缩干。加入150ml的水和150ml二氯甲烷,搅拌0.5h,静置分层。有机层浓缩干,得到18.9g油状液体产物2-氯-4-新戊基吡啶,收率85.1%。
[0047]1hnmr(400mhz,cdcl3):δppm 8.24(d,j=8.0hz,1h),7.09(d,j=8.0hz,1h),6.97(s,1h),2.46(s,2h),0.91(s,9h)。
[0048]
实施例3
[0049]
步骤(1):将4-吡啶甲醛(40g)室温溶于乙酸乙酯(400ml),体系搅拌溶清,降温并控温-50~-40℃,滴加1.7m叔丁基溴化镁(300ml),滴加完毕。自然升温至40-50℃,反应4h。反应结束后降温至0-10℃,滴加饱和氯化铵溶液淬灭反应,搅拌1h,静置分层,水层用乙酸乙酯萃取一次,合并有机层50℃浓缩干,得到48.1g淡黄色固体产物4-(2-羟基新戊基)吡啶,收率81.1%。
[0050]
步骤(2):将上一步得到的产品(25g)和10%钯碳(2.5g)、甲醇(250ml)加至反应瓶,降温至(0-10℃),通入氢气。反应20h,原料反应完全,过滤,无水硫酸钠干燥。有机层浓缩至干。得到17.8g油状液体产物4-新戊基吡啶,收率78.6%。
[0051]
步骤(3):将上一步得到的产品(10g)和甲醇100ml、双氧水(10ml)加至反应瓶,升温至50-60℃反应10h,反应完全,浓缩甲醇,降至室温,加入二氯甲烷,搅拌,静置分层。有机层浓缩干,得到8.79g油状液体产物4-新戊基吡啶氧化物,收率79.5%。
[0052]
步骤(4):将上一步得到的产品(5g)和三氯氧磷(50ml)加至反应瓶,升温回流反应5h。反应完全,浓缩干。加入15ml的水和15ml二氯甲烷,搅拌0.5h,静置分层。有机层浓缩干,得到4.4g油状液体产物2-氯-4-新戊基吡啶,收率80.1%。
[0053]1hnmr(400mhz,cdcl3):δppm 8.24(d,j=8.0hz,1h),7.09(d,j=8.0hz,1h),6.97(s,1h),2.46(s,2h),0.91(s,9h)。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips