
 商标分类
商标分类  商标转让
商标转让 
一种高纯度盐酸普拉克索的制备方法与流程
 2021-02-02 00:02:48|
2021-02-02 00:02:48| 437|
437| 起点商标网
起点商标网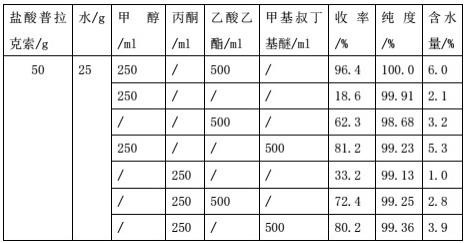
[0001]
本发明属于医药制剂领域,具体涉及一种高纯度盐酸普拉克索的制备方法。
背景技术:
[0002]
盐酸普拉克索是一种治疗帕金森综合征药物,属选择性非麦角类多巴胺受体激动剂,由德国勃林格殷格翰(boehringeringelheim)公司研发成功,是目前全球处方用量最大的多巴胺受体激动剂,1997年5月首次获得美国fda批准用于特发性帕金森病的治疗,能减轻患者的症状和体征,可以单独或与左旋多巴联合应用,在美国上市销售的商品名mirapex,是fda在过去6年中首次批准用于帕金森的药物,2007年在中国上市,商品名森福罗,用来治疗特发性帕金森病的体征和症状,单独(无左旋多巴)或与左旋多巴联用。
[0003]
盐酸普拉克索化学名为s(-)-2-氨基-6-正丙氨基-4,5,6,7-四氢苯并噻唑二盐酸盐,目前,它的合成方法只要有:1、文献j.med.chem.1987,30-494-498公开了一种盐酸普拉克索的制备方法,该方法以(-)-2,6-二氨基-4,5,6,7-四氢苯并噻唑为原料,在三乙胺的四氢呋喃中与丙酸酐反应,所得溶液用乙酸乙酯提取,经mgso4干燥、减压蒸除、丙酮洗涤,得到(-)-2-氨基-6-丙酰胺基-4,5,6,7-四氢苯并噻唑,在四氢呋喃中并通n2保护下,与硼烷的四氢呋喃溶液反应,游离碱用乙酸乙酯精制,在甲醇中成盐,成盐后再用甲醇精制一次得到盐酸普拉克索。
[0004]
该方法的缺点在于硼烷为无色剧毒气体,易燃、易爆、易水解,稳定性差,不易保存运输,反应安全性低,该方法制得的盐酸普拉克索收率低且没有结晶水,另外,硼烷的四氢呋喃溶液制造方法复杂,因此不适合工业化生产。
[0005]
2、专利cn201710706979.8的制备方法为:将2.11g(s)-(-)-2-氨基-6-正丙氨基-4,5,6,7-四氢苯并噻唑,然后加入30ml丙酮。冰浴降温至0~5℃,然后滴加3ml浓盐酸,控制在半小时内滴完。滴完后继续保温反应1小时。过滤,用10ml丙酮洗涤滤饼。滤饼在60℃下鼓风干燥1小时得到 2.53g普拉克索粗品。
[0006]
先加入15ml 85%(w/w)乙醇水溶液,升温至回流,共加入17ml 85%(w/w)乙醇水溶液使其全部溶清。加入0.3g活性炭,保温搅拌半小时。热滤。滤液先自然冷却至35℃析出晶种,然后继续在35℃保温析晶1小时。再用冰浴冷却至0℃,继续0℃保温析晶1小时。抽滤,用10ml无水乙醇洗涤滤饼。然后在50℃下鼓风干燥2小时,得到2.27g白色粉末状固体,hplc 检测含量99.95%,收率为75.17%。
[0007]
该方法涉及到成盐及精制,采用乙醇-水精制,高纯度,但是收率较低。
[0008]
3、专利 cn201310105277.6的制备方法为:将40.00g起始原料投入到1000ml的三口反应瓶中,加入160.0ml四氢呋喃,再加入19.85g碳酸氢钠,搅拌升温到50℃,滴加29.22g丙酸酐,控制在1小时滴加完毕,滴加完毕后搅拌反应 0.5小时,之后降温到5
±
5℃,再加入26.82g硼氢化钠,滴加三氟化硼乙醚溶液118.4ml,控制在1小时滴加完毕,滴加完毕后保温搅拌1小时,自然升到室温搅拌1.5小时,回流搅拌反应1.5小时;降温到10
±
5℃,滴加水120.0ml,20 分钟后加入100.0ml浓盐酸,加热回流1小时,冷却到15
±
5℃,用氢氧化钠溶液
调ph到大于 11,分层,水层再用适量乙酸乙酯萃取2次,合并有机层,用饱和盐水洗涤3次,再向有机层中加入150ml饱和盐水,搅拌下滴加盐酸16ml,使产物成一盐酸的盐酸盐析出,过滤析出的固体,60℃干燥后得到普拉克索一盐酸盐35.25g,收率60.20%, hplc含量大于99.8%。
[0009]
取以上得到的固体20.0g加入反应瓶中,加入100ml的无水乙醇,滴加浓盐酸13.5ml,搅拌反应30min,蒸溜去除溶剂及多余的酸,再加入8ml水和60ml无水乙醇加热溶清,热过滤,再加入60ml无水乙醇,搅拌降温析晶,在5-15 ℃温度下析晶2小时,过滤,干燥后得到普拉克索二盐酸一水合物21.50g,收率为87.8%, hplc含量99.9%。
[0010]
该方法先成一盐酸盐,再成二盐酸盐水合物,过程繁琐,总收率较低。
[0011]
4、专利 cn201310684092.5的制备方法为:(1)(s)-(-)-2-氨基-6-丙氨基-4,5,6,7-四氢苯并噻唑的制备将(s)-(-)-2,6-二氨基-4,5,6,7-四氢苯并噻唑(42.3g,0.25mol)加至甲醇(500ml)中,搅拌冷却至-20℃,控温在-15~-20℃滴加正丙醛(26.75g,0.46mol)。加毕,在该温度下搅拌反应1.5小时,控温-15~-20℃滴加nabh4(6.65g,0.18mol)的甲醇溶液80ml,加毕在自然温度下搅拌反应2小时。然后冷却至0℃以下,滴加浓盐酸70ml调节ph=2~3,然后蒸发掉甲醇,将残余物溶于150ml水中,用25%氢氧化钠溶液调节ph=7,于0℃下搅拌析晶2小时,过滤得白色鳞片状固体,用乙醇/水(体积比5:1)的混合溶剂精制两次,鼓风干燥得白色粉末状固体(s)-(-)-2-氨基-6-丙氨基-4,5,6,7-四氢苯并噻唑(33.5g,63.5%)。
[0012]
(2)普拉克索二盐酸盐一水合物的制备将(1)中得到的(s)-(-)-2-氨基-6-丙氨基-4,5,6,7-四氢苯并噻唑(33.5g,0.16mol)加至丙酮(300ml)中,-5~0℃搅拌下滴加浓盐酸13ml调节ph=1,然后于该温度下搅拌反应2 小时。过滤,滤饼用丙酮洗涤,得白色粉末状固体,然后用90%乙醇重结晶,50℃鼓风干燥得到普拉克索二盐酸盐一水合物(43.6g,90.3%)。
[0013]
该方法用丙酮做溶剂,浓盐酸成盐,收率低,需多次精制。
[0014]
现有技术中盐酸普拉克索合成方法,存在以下一些缺点:1)反应条件苛刻,反应安全性低,需要在惰性气体保护下进行,后处理复杂,需要除去溶剂,造成产物收率较低;2)需要多次重结晶,收率低,中间处理工艺较复杂,过程繁琐,不适于工业化生产;3)反应收率较低,纯度不高;4)采用丙酮和乙醇等单一的极性溶剂重结晶会造成结晶水流失,导致水份不合格。
[0015]
鉴于现有工艺的不足之处,十分必要开发一种反应步骤简单,安全性高,且收率高,纯度高,水份稳定,同时适合工业生产的盐酸普拉克索的新合成方法。
技术实现要素:
[0016]
本发明申请人发现,在普拉克索成盐及精制过程中,通过水、醇类试剂、酯类试剂三元体系精制,得到的盐酸普拉克索相较于一元或二元体系精制得到的盐酸普拉克索,收率高,纯度高,水分稳定,三元体系中水的比例提高,收率会显著的降低,醇类和酯类比例过高,结晶水的含量会逐步降低,不符合药典标准。
[0017]
本发明公布了一种高纯度盐酸普拉克索的制备方法,所述盐酸普拉克索的制备方法如下:
(a)以(s)-2-氨基-6-丙酰胺基-4,5,6,7-四氢苯并噻唑为原料,四氢呋喃做溶剂,在还原剂存在的条件下进行反应,反应完成后,加入稀盐酸终止反应;(b)加入氢氧化钠溶液,调节ph至12-14,加入萃取剂进行萃取,然后再加入醇类试剂,滴加浓盐酸成盐,降温析晶,过滤烘干后得到普拉克索二盐酸盐一水合物。
[0018]
(c)将步骤(b)得到的普拉克索二盐酸盐一水合物经过水、醇类试剂、酯类试剂三元体系精制,过滤,烘干得到盐酸普拉克索成品。
[0019]
进一步的,所述步骤(a)中的还原剂为硼氢化钠-三氟化硼乙醚或硼氢化钠-硫酸中的任一种,优选硼氢化钠-三氟化硼乙醚。
[0020]
进一步的,所述步骤(b)中的萃取剂为乙酸乙酯、乙酸异丙酯、甲基叔丁基醚中的任一种,优选乙酸乙酯,所述步骤(b)和步骤(c)中的醇类试剂为甲醇、乙醇、异丙醇中的任一种,优选甲醇,所述步骤(c)中的酯类试剂为乙酸乙酯、乙酸异丙酯中的任一种,优选乙酸乙酯。
[0021]
进一步的,所述步骤(a)中(s)-2-氨基-6-丙酰胺基-4,5,6,7-四氢苯并噻唑:四氢呋喃的体积比为:1:3-10。
[0022]
进一步的,所述步骤(b)中(s)-2-氨基-6-丙酰胺基-4,5,6,7-四氢苯并噻唑:醇类试剂的体积比为1:0.5-10。
[0023]
进一步的,所述步骤(b)中(s)-2-氨基-6-丙酰胺基-4,5,6,7-四氢苯并噻唑:浓盐酸的摩尔比为1:2-4,所述步骤(b)中析晶温度为0-20℃。
[0024]
进一步的,所述步骤(c)中(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑:水:醇类试剂:酯类试剂的体积比为:1:0:1:1-1:3:10:30。
[0025]
进一步的,所述步骤(a)中(s)-2-氨基-6-丙酰胺基-4,5,6,7-四氢苯并噻唑:硼氢化钠:三氟化硼乙醚的摩尔比为:1:2:2.4-1:4:4.8。
[0026]
进一步的,所述步骤(a)中(s)-2-氨基-6-丙酰胺基-4,5,6,7-四氢苯并噻唑:硼氢化钠:硫酸的摩尔比为:1:3:1.2-1:8:5。
[0027]
具体工艺过程如下:反应式:本发明的盐酸普拉克索的新合成方法反应步骤简单,安全性高,且收率高,纯度达到99.95%以上,水份稳定,同时适合工业生产。
附图说明
[0028]
图1为实施例1中盐酸普拉克索液相色谱图图2为实施例5中盐酸普拉克索液相色谱图图3为实施例7中盐酸普拉克索液相色谱图
具体实施方式
[0029]
以下通过实施例进一步说明本发明,但不作为对本发明的限制。
[0030]
实施例1将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃300ml、硼氢化钠20g加入反应瓶。控温10摄氏度滴加三氟化硼94.5g,滴完后升温至回流反应2小时。降至室温后加入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入乙酸乙酯萃取,分层。有机层浓缩至剩余4~5v溶剂后加入2v甲醇,滴加0.75v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物60.3g,摩尔收率89.9%,纯度99.92%。
[0031]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入25g水、250ml甲醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入500ml乙酸乙酯析晶。过滤,烘干得48.2g,水份:6.0%,收率96.4%,纯度100%。
[0032]
实施例2将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃400ml、硼氢化钠24g加入反应瓶。控温10摄氏度滴加三氟化硼113.4g,滴完后升温至回流反应2小时。降至室温后加入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入乙酸乙酯萃取,分层。有机层浓缩至剩余4~5v溶剂后加入2v甲醇,滴加0.8v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物61.5g,摩尔收率91.7%,纯度99.91%。
[0033]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入15g水、150ml甲醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入300ml乙酸乙酯析晶。过滤,烘干得48.5g,水份:6.0%,收率97%,纯度99.99%。
[0034]
实施例3将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃500ml、硼氢化钠22g加入反应瓶。控温10摄氏度滴加三氟化硼104g,滴完后升温至回流反应2小时。降至室温后加入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入乙酸乙酯萃取,分层。有机层浓缩至剩余4~5v溶剂后加入2v甲醇,滴加0.8v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物61.1g,摩尔收率91.1%,纯度99.91%。
[0035]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入5g水、100ml甲醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入200ml乙酸乙酯析晶。过滤,烘干得48.8g,水份:5.9%,收率97.6%,纯度99.98%。
[0036]
实施例4将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃500ml、硼氢化钠32g加入反应瓶。控温10摄氏度滴加三氟化硼104g,滴完后升温至回流反应2小时。降至室温后加入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入乙酸乙酯萃取,分层。有机层浓缩至剩余4~5v溶剂后加入5v甲醇,滴加1.4v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物61g,摩尔收率90.9%,纯度99.91%。
[0037]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入5g水、100ml甲醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入200ml乙酸乙酯析晶。过滤,烘干得48.7g,水份:5.9%,收率97.4%,纯度99.98%。
[0038]
实施例5将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃300ml、硼氢化钠40g加入反应瓶。控温10摄氏度滴加硫酸50g,滴完后升温至回流反应2小时。降至室温后加
入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入乙酸乙酯萃取,分层。有机层浓缩至剩余4~5v溶剂后加入2v甲醇,滴加0.75v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物60g,摩尔收率89.4%,纯度99.92%。
[0039]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入25g水、250ml乙醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入500ml乙酸乙酯析晶。过滤,烘干得48.3g,水份:6.0%,收率96.6%,纯度99.97%。
[0040]
实施例6将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃350ml、硼氢化钠41g加入反应瓶。控温10摄氏度滴加硫酸55g,滴完后升温至回流反应2小时。降至室温后加入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入乙酸异丙酯萃取,分层。有机层浓缩至剩余4~5v溶剂后加入2v甲醇,滴加0.75v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物60.5g,摩尔收率90.2%,纯度99.91%。
[0041]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入25g水、250ml异丙醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入500ml乙酸异丙酯析晶。过滤,烘干得48.5g,水份:6.1%,收率97.0%,纯度99.98%。
[0042]
实施例7将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃360ml、硼氢化钠42g加入反应瓶。控温10摄氏度滴加硫酸54g,滴完后升温至回流反应2小时。降至室温后加入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入甲基叔丁基醚萃取,分层。有机层浓缩至剩余4~5v溶剂后加入2v甲醇,滴加0.75v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物60.8g,摩尔收率90.6%,纯度99.93%。
[0043]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入25g水、250ml甲醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入500ml乙酸乙酯析晶。过滤,烘干得48.8g,水份:5.9%,收率97.6%,纯度99.97%。
[0044]
实施例8将(s)-2-氨基-6-丙酰胺基
-ꢀ
4,5,6,7-四氢苯并噻唑50g,四氢呋喃400ml、硼氢化钠60g加入反应瓶。控温10摄氏度滴加硫酸95g,滴完后升温至回流反应2小时。降至室温后加入稀盐酸终止反应。加入20%的氢氧化钠水溶液,调节ph至14后加入乙酸乙酯萃取,分层。有机层浓缩至剩余4~5v溶剂后加入6v甲醇,滴加1.0v浓盐酸成盐,降温至10℃析晶。过滤烘干后得到盐酸普拉克索二盐酸盐一水合物60.3g,摩尔收率89.9%,纯度99.92%。
[0045]
将50g盐酸普拉克索二盐酸盐一水合物加入反应瓶,加入25g水、400ml甲醇搅拌均匀后升温溶解,过滤除去机械杂质,缓慢加入800ml乙酸乙酯析晶。过滤,烘干得48.8g,水份:5.9%,收率97.6%,纯度99.97%。
[0046]
本发明申请人发现,在普拉克索成盐及精制过程中,通过水、醇类试剂、酯类试剂三元体系精制,得到的盐酸普拉克索收率高,纯度高,水分稳定,三元体系中水的比例提高,收率会显著的降低,醇类和酯类比例过高,结晶水的含量会逐步降低,不符合药典标准。三元体系相对于一元或二元体系精制,得到的盐酸普拉克索的收率、纯度高,盐酸普拉克索的含水量更稳定。
[0047]
盐酸普拉克索精制所用溶剂得到的纯度,收率及含水量对比如下:
通过以上试验对比可知,盐酸普拉克索在水和醇、水和酯的二元体系中精制时,收率比较低,含水量不符合要求,在与丙酮或甲基叔丁基醚的体系中精制时,收率与含水量效果不佳,盐酸普拉克索在水、醇、酯的三元体系中精制时得到的成品含量、纯度都比较高,含水量符合药典要求。
[0048]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips