
 商标分类
商标分类  商标转让
商标转让 
一种抗肿瘤活性熊果酸衍生物及其制备方法与流程
 2021-02-01 23:02:15|
2021-02-01 23:02:15| 510|
510| 起点商标网
起点商标网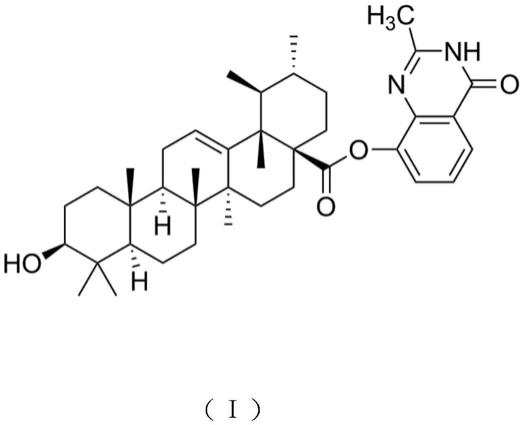
[0001]
本发明属于化学药物合成技术领域,涉及一种抗肿瘤活性熊果酸衍生物及其制备方法。
背景技术:
[0002]
恶性肿瘤是当今社会威胁人类的身体健康威胁最严重的疾病,它是100多种相关疾病的统称,其具有早起转移、晚期难治、预后差、易产生耐药性等特点。目前对早期或较早期实体肿瘤来说,手术切除仍然是首选的治疗方法,再辅助放、化疗等方法控制癌细胞扩散。但术后复发,放、化疗所带来的巨大的毒副作用使患者痛不欲生。为了减少患者的痛苦,延长患者生命,提高患者生活质量,人们一直在寻找高效低毒的抗肿瘤药物,尽管现有的抗肿瘤药物可以治疗并治愈部分实体瘤,但这类药物存在治疗周期长、毒副作用大、易产生耐药性等缺陷,部分抗肿瘤药物价格昂贵。因此,寻找高效、低毒、经济适用的抗肿瘤药物成为当务之急。
[0003]
现有的药理研究和临床研究表明,天然植物的提取物在治疗肿瘤疾病方面表现出了显著的优势,与常规的药物相比,其毒副作用小,见效快。熊果酸又名乌苏酸或乌索酸,属于三萜类化合物,广泛存在于中草药、食物等中,具有较好的抗癌活性,其抗癌主要表现在抗突变、抑制癌细胞启动、可直接杀伤癌细胞,具有肿瘤逆转作用及诱导癌细胞凋亡,从而抑制癌变形成。作为传统的中药,虽然熊果酸在抗肿瘤方面具有很好的优势,但由于熊果酸不溶于水,不能将其制备成液体制剂,致使熊果酸的药理活性无法得到充分利用。
技术实现要素:
[0004]
本发明提供了一种抗肿瘤活性熊果酸衍生物,其具有式(i)分子结构:
[0005][0006]
本发明的另一个目的在于提供一种抗肿瘤活性熊果酸衍生物的制备方法,包括如下步骤:
[0007]
(1)将熊果酸溶解在有机溶剂a中,在低温状态下缓慢加入氯化亚砜,滴加速度以控制反应体系的温度不超过0℃为准,加入2~3滴dmf(n,n-二甲基甲酰胺),调节反应温度为35~45℃,回流,反应结束,去除多余的氯化亚砜;
[0008]
(2)将步骤(1)产物溶解在有机溶剂b中,加入2-甲基-8-羟基-4-喹唑啉酮,在室温下回流反应,反应结束,依次加入5%的盐酸、蒸馏水、饱和食盐水洗涤有机相,无水硫酸钠干燥有机相1~3h,产物经硅胶柱层析分离,收集洗脱液,旋转蒸发除去石油醚和二氯甲烷,固体干燥得熊果酸衍生物;
[0009]
步骤(1)中,所述有机溶剂a为乙醚、四氢呋喃或二氯甲烷;
[0010]
所述熊果酸与氯化亚砜的投料摩尔比为1:5~10;
[0011]
步骤(2)中,所述有机溶剂b为二氯甲烷、氯仿、苯或甲苯;
[0012]
所述硅胶柱层析柱的填充剂为200~400目的硅胶;
[0013]
所述硅胶柱层析中的洗脱液为石油醚和二氯甲烷的混合液,两者的体积比为3:1~10:1。
[0014]
根据上述制备方法的一种优选,包括如下步骤:
[0015]
(1)将熊果酸溶解在四氢呋喃中,在低温状态下缓慢加入氯化亚砜,滴加速度以控制反应体系的温度不超过0℃为准,加入2~3滴dmf(n,n-二甲基甲酰胺),调节反应温度为40~45℃,回流,反应结束,去除多余的氯化亚砜;
[0016]
(2)将步骤(1)产物溶解在二氯甲烷中,加入2-甲基-8-羟基-4-喹唑啉酮,在室温下回流反应,反应结束,依次加入5%的盐酸、蒸馏水、饱和食盐水洗涤有机相,无水硫酸钠干燥有机相3h,产物用200~300目硅胶拌样,样品过硅胶柱层析分离(洗脱剂:v
石油醚
:v
二氯甲烷
=3:1),收集洗脱液,旋转蒸发除去石油醚和二氯甲烷,固体干燥得熊果酸衍生物。
[0017]
与现有技术相比,本发明的有益效果是:
[0018]
(1)本发明将熊果酸与2-甲基-8-羟基-4-喹唑啉酮结合,制备结构新颖的熊果酸衍生物,引入的喹唑啉基团增加了新化合物的水溶性;病理实验表明,熊果酸衍生物可以抑制部分肿瘤细胞生长,尤其对卵巢癌细胞(a2780)、人胃癌细胞(sgc-7901)及人肝癌细胞(hepg2)具有有显著的抑制活性;
[0019]
(2)本发明所提供的制备方法简单、原料易得、收率高、操作条件温和,可以方便地制备出结构新颖的熊果酸衍生物,适合工业化生产。
附图说明
[0020]
图1:实施例1的抗肿瘤活性熊果酸衍生物的核磁共振氢谱图。
具体实施方式
[0021]
为了使本发明的目的、技术方案及优点更加清楚明白,下面结合具体实施方式对本发明作进一步的详细描述,但不应将此理解为本发明上述主题的范围仅限于下述实施例。
[0022]
实施例1
[0023]
(1)将25g熊果酸溶解在50ml四氢呋喃中,缓慢滴加氯化亚砜19.9ml,滴加速度以控制反应体系的温度不超过0℃为准,滴毕后搅拌15~30min,加入2~3滴dmf(n,n-二甲基
甲酰胺),调节反应温度为40~45℃,回流反应3h,反应结束,去除多余的氯化亚砜;
[0024]
(2)将15g步骤(1)产物溶解在100ml二氯甲烷中,加入7.6g 2-甲基-8-羟基-4-喹唑啉酮,在室温下回流反应3h,反应结束,依次加入5%的盐酸、蒸馏水、饱和食盐水洗涤有机相,有机相再用无水硫酸钠除水3h,将干燥后的产物加入无水乙醇溶解,再用200~300目硅胶拌样,样品过硅胶柱层析分离(洗脱剂:v
石油醚
:v
二氯甲烷
=3:1),收集洗脱液,旋转蒸发除去石油醚和二氯甲烷,固体干燥得熊果酸衍生物,产率为82.13%。
[0025]
核磁共振氢谱检测:
[0026]
将样品放入样品管中,用注射器取0.5mlcdcl3(氘代氯仿)注入样品管,使样品充分溶解。要求样品与试剂充分混合,溶液澄清、透明、无悬浮物或其他杂质,经核磁共振鉴定,得到核磁共振氢谱图,结果见如图1。
[0027]
实施例2本发明化合物的水溶性测试
[0028]
称取0.1g实施例1化合物于试管中,加入纯化水10ml,室温下每隔5min振摇30秒,30min后观察溶解情况,记录溶剂用量,将实验结果折算为标准溶解度(25℃),测试结果见表1。
[0029]
表1实施例1化合物溶解度与收率
[0030][0031]
结果表明,与熊果酸相比,本发明的熊果酸衍生物的水溶性有显著提高。
[0032]
实施例3本发明的化合物的抗肿瘤活性测试
[0033]
对本发明化合物进行了肿瘤细胞增殖抑制试验,试验方法采用常规的mtt法。
[0034]
细胞株选用:乳腺癌细胞(mcf-7)、卵巢癌细胞(a2780)、人肺癌细胞(a-549)、人宫颈癌细胞(hela)、人胃癌细胞(sgc-7901)、人肝癌细胞(hepg2)。培养液为dmem+15%nbs+双抗。
[0035]
样品液的配制:用dmso(merck)将实施例1化合物溶解后,加入pbs(-)配成100umol/l的溶液或者均匀的混悬液,然后用dmso的pbs(-)稀释,最终浓度为0.1,1,10,20,40,60,80,100umol/l。
[0036]
以同样的条件配制2-甲基-8-羟基-4-喹唑啉酮(化合物a)溶液与熊果酸溶液,将配制的化合物a溶液作为对照组2,熊果酸溶液作为对照组3。
[0037]
以同样的条件配制抗肿瘤药物紫杉醇为对照品溶液(对照组1)。
[0038]
细胞培养:贴壁生长肿瘤细胞培养于含10%灭活新生牛血清和青霉素、链霉素(各100万u/l)的1640培养液中,置于37℃,饱和co2培养箱中培养。细胞贴壁生长,每2~3天传代一次,传代时首先倒出培养液,pbs洗2次,胰酶消化后,加入新鲜的培养液吹打均匀,调整细胞至适当浓度移入新的培养瓶中,添加培养液至适量,取对数生长期细胞用于实验。
[0039]
mtt法检测细胞活性及ic
50
的测定:
[0040]
实验原理:活细胞线粒体中的琥珀酸脱氢酶能使外源性mtt还原为水不溶性的蓝
紫色结晶甲瓒(formazan)并沉积在细胞中,而死细胞无此功能。二甲基亚砜(dmso)能溶解细胞中的甲瓒,用酶联免疫检测仪在490nm波长处测定其光吸收值,可间接反映活细胞数量。
[0041]
实验方法:取对数生长期的细胞,消化、计数,以2
×
104/ml的密度接种于96孔培养板中,每孔100ul。培养24小时后,将待测化合物以0.1,1,10,20,40,60,80,100浓度处理细胞。实验组每个浓度设5个复孔,以含0.4%dmso的培养液作为对照。药物作用48h后,去上清,每孔加入100ulmtt(2-(4,5-二甲基-2-噻唑基)-3,5-二苯基-2h-四唑氢溴酸盐)(1mg/ml),继续培养4h,弃上清液,每孔加入100ul dmso,振荡摇匀,用酶标仪在570nm处测定吸光度值,采用ic
50
计算软件求出半数抑制浓度(ic50),试验结果见表2。
[0042]
表2本发明化合物对不同肿瘤细胞的半数抑制浓度ic
50
[0043][0044]
试验结果表明,本发明合成的熊果酸衍生物可以抑制肿瘤细胞生长,尤其对卵巢癌细胞(a2780)、人胃癌细胞(sgc-7901)及人肝癌细胞(hepg2)有显著的抑制活性,说明本发明的化合物具有开发抗肿瘤药物的前景和意义。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips