
 商标分类
商标分类  商标转让
商标转让 
一种制备抗真菌药硫酸艾沙康唑鎓的方法与流程
 2021-02-01 23:02:28|
2021-02-01 23:02:28| 230|
230| 起点商标网
起点商标网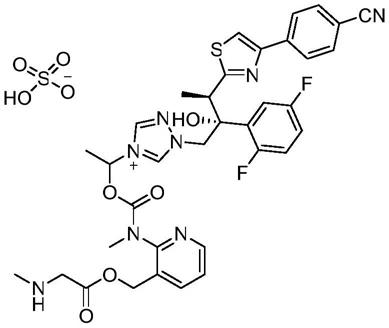
[0001]
本发明涉及制备抗真菌药物硫酸艾沙康唑鎓的方法。
背景技术:
[0002]
硫酸艾沙康唑鎓(cresemba)是一种新型三唑类抗真菌药,2015年3月在美国批准上市。该药由瑞士巴塞利来和日本安斯泰来公司联合开发,用于治疗侵袭性曲霉病和侵袭性毛霉菌病,其化学式如下所示:
[0003][0004]
目前,由于广谱抗生素、激素的广泛应用,加剧了真菌病的蔓延,使深部真菌感染成为癌症病人、aids病人及免疫丧失病人死亡的因素之一,其中唑类抗真菌药物是目前临床应用的一线药物,对浅部真菌和深部真菌都具有明显的治疗效果。由于第一代抗真菌唑类药-氟康唑、伊曲康唑和第二代抗深部真菌药伏立康唑、泊沙康唑面临一些问题,如:多药耐药、新致病菌的出现等,此限制了这几种药物的临床广泛应用,因此需要进一步开发新的用于预防和治疗深部真菌感染的药物。其中艾沙康唑为新一代三唑类抗真菌药,其水溶性前药硫酸艾沙康唑鎓的设计不仅解决了水溶性的问题,且还减小了毒副作用,为临床治疗曲霉病、念球菌及其它少见真菌感染提供新的选择。
[0005]
在硫酸艾沙康唑鎓的合成中,实现由艾沙康唑鎓季铵盐的卤素转变为硫酸氢盐的盐型转化是工艺的技术难点,由于艾沙康唑鎓具有热不稳定性、结构中含多成盐位点等因素,更增加了合成难度。目前,原研公司的专利cn1185230c中并没有公开盐型转化的工艺方法,对于该盐型转化的工艺方法也鲜有报道。霍彩霞等(国际药学研究杂志,2016年,第43卷第3期,436-440页)采用羟基型离子交换树脂的方法制备氢氧化艾沙康唑鎓,进而采用硫酸实现盐型转化,合成路线如下所示。经实验发现,氢氧化艾沙康唑鎓的稳定性极差,工艺可操作性不强,很难实现工业化生产。
[0006][0007]
扬子江药业的专利cn106916152a报道了通过氧化还原反应制备艾沙康唑单硫酸盐,合成路线如下所示。该方法需使用柱层析纯化,增加了成本,也难以进行工业化生产。此外,由于艾沙康唑鎓存在多个碱性氮,理论上难以直接和硫酸成盐得到单硫酸盐。
[0008][0009][0010]
专利2016/016766a2报道了将化合物5在甲醇中与离子交换树脂(hso
4﹣
形式)一步
置换得到产物1,实验路线如下所示。实验研究发现,存在以下问题:一方面,离子交换树脂很难将化合物5中的氯化氢分子置换完全,即使是使用过量的离子交换树脂;另一方面,离子交换过程在甲醇中进行,产物1对甲醇不稳定,不仅发生降解产生多个杂质,且终产品难以进行精制纯化。因此,该工艺方法难以得到合格的目标产物。
[0011][0012]
因此,需要进行艾沙康唑鎓卤代盐或者艾沙康唑鎓氢氧化物转变为硫酸氢盐的制备工艺研究,以解决盐型转化的技术难点,得到合格的硫酸艾沙康唑鎓样品,并实现该产品的规模化制备。
技术实现要素:
[0013]
本发明的目的是公开一种制备抗真菌药硫酸艾沙康唑鎓的方法,以克服现有技术存在的缺陷。
[0014]
本发明的方法,包括如下步骤:
[0015]
将式6化合物在有机溶剂水溶液中,与苯乙烯系硫酸氢阴离子交换树脂交换离子后,分液,然后从水相中,收集所述的硫酸艾沙康唑鎓(1);
[0016]
所述的苯乙烯系硫酸氢阴离子交换树脂,是采用强碱性苯乙烯系阴离子交换树脂和硫酸氢盐溶液制备的,制备方法如下:
[0017]
将强碱性苯乙烯系阴离子交换树脂与硫酸氢盐溶液混合后浸泡2~6h,然后,过滤,洗涤树脂至滤液为中性,即可获得所述的苯乙烯系硫酸氢阴离子交换树脂;
[0018]
所述的硫酸氢盐溶液的摩尔浓度为0.5~2mol/l;
[0019]
反应通式如下:
[0020]
[0021]
离子树脂的体积(ml)与化合物6的质量(g)的比值为20.4/a~34.1/a,其中,a为离子交换树脂的交换容量,单位为mmol/ml;
[0022]
所述的离子交换树脂为阴离子交换树脂,优选地,为强碱性苯乙烯系阴离子交换树脂,可采用商业化产品,如阿拉丁公司型号为719的产品;
[0023]
所述的有机溶剂为二氯甲烷、乙酸乙酯或乙酸异丙酯中的一种以上,优选地,为二氯甲烷,水和有机溶剂的体积比为1:150~1:200,优选地,为1:160~1:170;
[0024]
反应温度为15℃~30℃,优选地,为20℃~25℃,反应时间为1~2h,优选地,为1.2h~1.5h;
[0025]
优选的,从水相中收集所述的硫酸艾沙康唑鎓(1)的方法,包括如下步骤:
[0026]
将水相采用有机溶剂洗涤,然后冻干,即可获得述的硫酸艾沙康唑鎓(1);
[0027]
所述的洗涤有机溶剂为二氯甲烷、乙酸乙酯或乙酸异丙酯,优选地,为二氯甲烷;
[0028]
所述的化合物6的制备方法,包括如下步骤:
[0029]
式6化合物的制备方法包括如下步骤:
[0030]
将式5化合物加入到有机溶剂水溶液中,然后加入无机碱溶液调节ph为6.5~8.5,优选的为7.5~8.0,分液,然后从反应产物中,收集所述的艾沙康唑鎓的氢氧化物(6),反应温度为-5℃~5℃,优选的为0℃~3℃;
[0031][0032]
所述的无机碱选自碳酸钠、碳酸钾、碳酸氢钠或碳酸氢钾中的一种或以上,优选地,为碳酸氢钠和碳酸氢钾,无机碱溶液的质量分数为2%~10%,优选地,为5%~6%;
[0033]
所述的有机溶剂为二氯甲烷、乙酸乙酯或乙酸异丙酯,优选地,为二氯甲烷;水和有机溶剂的体积比为1:0.8~1:1.2,优选地,为1:1~1:1.1;
[0034]
所述的式5化合物,可采用如下的路线进行制备:
[0035][0036]
式2化合物和式3化合物,可采用专利cn1185230c公开的方法制备;
[0037]
本发明的有益效果是:
[0038]
本发明使用苯乙烯系硫酸氢阴离子交换树脂实现艾沙康唑鎓的碘盐或氢氧化物转化为硫酸氢盐,此方法避免了多硫酸盐的形成,工艺操作简单,无需经柱层析纯化及重结晶操作,产物纯度达到药用要求,易于规模化制备。
[0039]
新的工艺具有如下技术特点:
[0040]
1)通过hso4离子负载的离子交换树脂与艾沙康唑鎓底物进行离子交换,实现艾沙康唑鎓的碘盐或氢氧化物转化为硫酸氢盐,避免了多硫酸盐的形成,该方法未见文献报道;
[0041]
2)苯乙烯系硫酸氢阴离子交换树脂由任何型号的强碱性苯乙烯系阴离子交换树脂与硫酸氢盐溶液制备,现制现用;
[0042]
3)针对终产物遇热不稳定,难以进行重结晶纯化的技术难题,可经离子交换后,水相冻干,无需进一步纯化来解决。
具体实施方式
[0043]
下面通过实例进一步描述本发明的技术方案,这些实施例为示范性的,不构成对本发明保护范围的限定。本领域的技术人员,在本发明的指导下,根据现有技术而对其中技术特征进行等同替换仍属于本发明的保护范围内。
[0044]
实施例1
[0045]
1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓盐酸盐碘代物(式5化合物)的制备:
[0046]
将式2化合物(100g)、式3化合物(133g)、nai(48g)以及无水乙腈(500ml)加入到三颈烧瓶中,氮气保护。搅拌下升温至内温70℃,保温搅拌3h。降温至20℃以下,过滤,滤饼用乙酸乙酯洗涤,合并,浓缩。向浓缩物中加入乙酸乙酯(400ml)和纯化水(1000ml),搅拌萃取。分液,上层有机层为酒红色,下层用乙酸乙酯(100ml)萃取一次,合并有机相,用饱和食盐水(100ml)洗涤,加入40g无水硫酸钠干燥,减压浓缩得到泡沫状固体,直接用于下一步反应。收率98%,hplc纯度93%。
[0047]
将式4化合物(100g)、无水乙酸乙酯(1000ml)加入到三颈烧瓶中,氮气保护。滴加2mol/l的氯化氢乙酸乙酯溶液(530ml),滴加完毕后继续搅拌2h。氮气保护下过滤,滤饼在真空下40℃干燥6h,得黄色固体,即获得化合物5。收率92%,hplc纯度94%。
[0048]
反应通式如下:
[0049][0050]
式2化合物和式3化合物,可采用专利cn1185230c公开的方法制备;
[0051]
实施例2
[0052]
1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓氢氧化物(化合物6)的制备:
[0053]
将式5化合物(50g)、二氯甲烷(2000ml)和去离子水(1600ml)加入到三颈烧瓶中,氮气置换,降温至0℃。滴加质量浓度为2%碳酸氢钠溶液,保持反应体系温度小于5℃,调节ph至6.5,分液,下层有机层加去离子水(500ml)洗涤一次,然后真空浓缩,即可获得所述的化合物6。
[0054]
hplc纯度90%。离子色谱检测oh-含量为12.34%。
[0055]
1h nmr(400mhz,dmso)δ10.280(m,1h),9.345(m,1h),9.108(s,2h),8.580(s,2h),8.486(s,2h),8.233(d,j=8.0hz,2h),7.946(m,3h),7.472(m,1h),7.355(m,1h),7.248(t,j=0.8hz,1h),7.060(d,j=0.8hz,1h),6.796(d,j=0.8hz,1h),6.638(m,1h),5.227(s,j=0.8hz,1h),5.005(m,2h),4.719(t,j=8.0hz,1h),4.175(m,1h),4.079(s,2h),3.200(d,j=16hz,3h),2.600(s,3h),1.695(m,3h),1.200(d,j=8hz,3h).
[0056]
es-ms m/z=717(m
+
)
[0057]
实施例3
[0058]
1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1)的制备:
[0059]
将离子交换树脂用去离子水洗、酸洗和碱洗后,加入浓度为1mol/l的硫酸氢钠溶液浸泡4h,过滤,树脂水洗至滤液为中性,制得苯乙烯系硫酸氢阴离子交换树脂。
[0060]
所述的离子交换树脂为强碱性苯乙烯系阴离子交换树脂,采用阿拉丁科技公司型号为719的产品,719离子交换树脂的交换容量为1.4mmol/ml;
[0061]
将45克化合物6加入到二氯甲烷1500ml和去离子水10ml的混合液中,氮气保护下,加入656ml上述的离子交换树脂,15℃下搅拌1h,分液,收集上层水层,加二氯甲烷50ml洗涤两次,冻干,得白色粉末,即为所述的1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1);
[0062]
化合物6的质量与离子交换树脂的体积的比值为14.5;
[0063]
总收率50%(以式2计),纯度达到药用样品要求。离子色谱检测so
42-含量为13.06%。1h nmr(400mhz,dmso)δ10.280(m,1h),9.345(m,1h),9.108(s,2h),8.580(s,2h),8.486(s,2h),8.233(d,j=8.0hz,2h),7.946(m,3h),7.472(m,1h),7.355(m,1h),7.248(t,j=0.8hz,1h),7.060(d,j=0.8hz,1h),6.796(d,j=0.8hz,1h),6.638(m,1h),5.227(s,j=0.8hz,1h),5.005(m,2h),4.719(t,j=8.0hz,1h),4.175(m,1h),4.079(s,2h),3.200(d,j=16hz,3h),2.600(s,3h),1.695(m,3h),1.200(d,j=8hz,3h).
[0064]
es-ms m/z=717(m
+
)。
[0065]
实施例4
[0066]
1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1)的制备:
[0067]
将719离子交换树脂按实施例3方式处理,备用。
[0068]
将45克化合物6加入到二氯甲烷1500ml和去离子水10ml的混合液中,氮气保护下,加入1096ml上述的离子交换树脂,15℃下搅拌1h,分液,收集上层水层,加乙酸乙酯50ml洗涤两次,冻干,得白色粉末,即为所述的1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1);
[0069]
化合物6的质量与离子交换树脂的体积的比值为47.7;
[0070]
总收率50%(以式2计),纯度达到药用样品要求。离子色谱检测so
42-含量为14.86%。1h nmr(400mhz,dmso)δ10.280(m,1h),9.345(m,1h),9.108(s,2h),8.580(s,2h),8.486(s,2h),8.233(d,j=8.0hz,2h),7.946(m,3h),7.472(m,1h),7.355(m,1h),7.248(t,j=0.8hz,1h),7.060(d,j=0.8hz,1h),6.796(d,j=0.8hz,1h),6.638(m,1h),5.227(s,j=0.8hz,1h),5.005(m,2h),4.719(t,j=8.0hz,1h),4.175(m,1h),4.079(s,2h),3.200(d,j=16hz,3h),2.600(s,3h),1.695(m,3h),1.200(d,j=8hz,3h).
[0071]
es-ms m/z=717(m
+
)。
[0072]
实施例5
[0073]
1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1)的制备:
[0074]
将719树脂按实施例3方式处理,备用。
[0075]
将45克化合物6加入到二氯甲烷2000ml和去离子水10ml的混合液中,氮气保护下,加入656ml上述的离子交换树脂,15℃下搅拌1h,分液,收集上层水层,加乙酸异丙酯50ml洗涤两次,冻干,得白色粉末,即为所述的1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1);
[0076]
化合物6的质量与离子交换树脂的体积的比值为14.6;
[0077]
总收率50%(以式2计),纯度达到药用样品要求。离子色谱检测so
42-含量为13.21%。1h nmr(400mhz,dmso)δ10.280(m,1h),9.345(m,1h),9.108(s,2h),8.580(s,2h),
8.486(s,2h),8.233(d,j=8.0hz,2h),7.946(m,3h),7.472(m,1h),7.355(m,1h),7.248(t,j=0.8hz,1h),7.060(d,j=0.8hz,1h),6.796(d,j=0.8hz,1h),6.638(m,1h),5.227(s,j=0.8hz,1h),5.005(m,2h),4.719(t,j=8.0hz,1h),4.175(m,1h),4.079(s,2h),3.200(d,j=16hz,3h),2.600(s,3h),1.695(m,3h),1.200(d,j=8hz,3h).
[0078]
es-ms m/z=717(m
+
)。
[0079]
实施例6
[0080]
1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1)的制备:
[0081]
将719离子交换树脂按实施例3方法处理,备用
[0082]
将45克化合物6加入到二氯甲烷1500ml和去离子水10ml的混合液中,氮气保护下,加入656ml上述的离子交换树脂,30℃下搅拌1h,分液,收集上层水层,加二氯甲烷50ml洗涤两次,冻干,得白色粉末,即为所述的1-[3-(r)-[4-(4-氰基苯基)噻唑-2-基]-(2r)-(2,5-二氟苯基)-2-羟基丁基-4-[1-[n-甲基-n-[3-[2-(甲氨基)乙酰氧基甲基]吡啶-2-基]氨基甲酰氧基]乙基]-1h-1,2,4-三唑鎓硫酸盐(化合物1);
[0083]
化合物6的质量与离子交换树脂的体积的比值为14.6;
[0084]
总收率50%(以式2计),纯度达到药用样品要求。离子色谱检测so
42-含量为13.66%。1h nmr(400mhz,dmso)δ10.280(m,1h),9.345(m,1h),9.108(s,2h),8.580(s,2h),8.486(s,2h),8.233(d,j=8.0hz,2h),7.946(m,3h),7.472(m,1h),7.355(m,1h),7.248(t,j=0.8hz,1h),7.060(d,j=0.8hz,1h),6.796(d,j=0.8hz,1h),6.638(m,1h),5.227(s,j=0.8hz,1h),5.005(m,2h),4.719(t,j=8.0hz,1h),4.175(m,1h),4.079(s,2h),3.200(d,j=16hz,3h),2.600(s,3h),1.695(m,3h),1.200(d,j=8hz,3h).
[0085]
es-ms m/z=717(m
+
)。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips