
 商标分类
商标分类  商标转让
商标转让 
一种化学合成酸性多肽的制备方法与流程
 2021-02-01 22:02:33|
2021-02-01 22:02:33| 401|
401| 起点商标网
起点商标网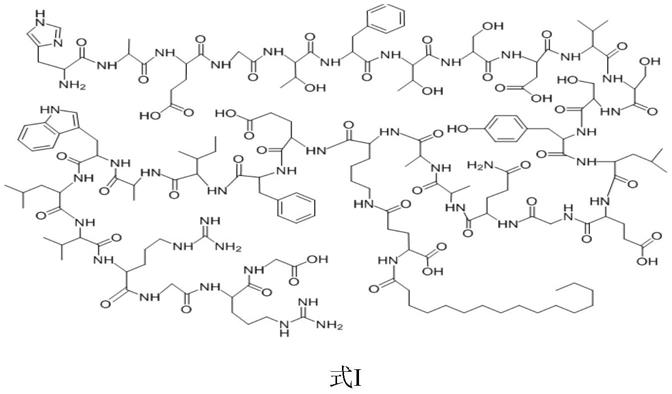
[0001]
本发明涉及一种化学合成酸性多肽的制备方法,特别涉及一种利用反相制备色谱对化学方法合成的酸性多肽化合物进行纯化。
技术背景
[0002]
化学合成的多肽化合物的生物活性极高,要求药用的多肽化合物,需要具有极高的化合物纯度,极低的杂质含量,以有效避免在临床上因杂质对患者引起的不适反应。在多肽的化学合成过程中会产生一系列的缺失、水解、消旋杂质,这些杂质在药物临床使用时因其毒性,或者其可能存在与期望多肽不符的生物活性,可能会造成药物诸多的毒副作用,而这些杂质与目标化合物的性质十分相近,难于分离,在工业制备中是非常严峻的考验。
[0003]
现阶段酸性多肽化合物,特别是分子量较大的、溶解性较差的弱酸性多肽在采用制备色谱进行分离纯化时,现有色谱柱填料大多对酸碱的耐受范围有限,采用碱性洗脱液进行洗脱时对色谱柱的损耗十分严重,而采用酸性洗脱液又很难达到分离出高纯度目标化合物的目的。酸性化合物在酸性溶液下,溶解度较碱性溶液小。如式ι所示的多肽由于其肽链长且侧链含有较长的烷基修饰、疏水氨基酸的存在导致疏水性非常强,其在酸性体系下溶解度非常小,使用酸性缓冲体系纯化难度非常大。
[0004]
在已有的文献中,如专利cn103275209a采用0.1%tfa/水溶液-0.1%tfa/乙腈溶液作为洗脱液,专利cn 201210029818.7采用,以含有0.1~0.2%三氟醋酸的异丙醇水溶液为a相,含有0.1~0.2%三氟醋酸的乙腈为b进行第一步纯化,专利cn201410001671.x采用a相:0.1%tfa;b相:乙腈作为第一次纯化条件,专利cn201810663478.0采用磷酸作为流动相。这些方法中大多采用醋酸、三氟乙酸、磷酸作为流动相的组成部分,但是因洗脱过程中样品吸附在填料上,导致回收率偏低等问题,不适合作为制备流动相体系;同时制备收集的馏分放置一段时间之后样品析出,很难再溶清,不适合工业化生产。也有文献报道采用碱性流动相进行纯化,但不可避免地需要采取特殊的色谱柱,涉及到的色谱柱子的填料品种较多并且有的填料非常昂贵。不适合大规模工业化生产。
技术实现要素:
[0005]
有鉴于此,本发明为解决现有技术中酸性多肽化合物,尤其是溶解度差、弱酸性化多肽化合物纯化杂质困难、收率低的缺点,提供了一种利用反相制备色谱对化学方法合成的酸性多肽化合物进行纯化的方法。
[0006]
为实现上述目的,本发明提供的技术方案如下:
[0007]
一种用于从包含所述酸性多肽和相关杂质的混合物中纯化制备高纯度酸性多肽的方法,所述方法是一种高效液相色谱法,分两步或三步进行rp-hplc纯化,用于洗脱的溶剂主要由离子对试剂和有机改性剂组成,所述洗脱溶剂以梯度洗脱的方式进行洗脱,
[0008]
其中,所述离子对试剂选自nh4cl,hcooh,nh4hco3,teap溶液;
[0009]
其中,所述有机改性剂选自甲醇、乙醇、正丙醇、异丙醇、乙腈。
[0010]
多肽化学合成中,杂质主要为反应过程中的副产物、中间体、降解产物、氧化产物、盐及各种保护基。其中,缺失肽和消旋肽为主要的需要去除杂质。
[0011]
本发明的一些实施例中,酸性多肽具有如下结构:
[0012][0013]
如式ι所示的酸性多肽,肽链较长、支链结构复杂,在合成过程中产生的杂质多,在作为高纯度要求的药物活性成分时,需要严格控制纯度,必须对合成的粗肽进行纯化。
[0014]
本发明所述的酸性多肽,尤其指肽序中含有疏水性较强的氨基酸、有较长的烷基侧链修饰而造成溶解性较差的弱酸性多肽。纯化过程中难以洗脱,造成损失,增加了纯化难度,影响了酸性多肽的纯化收率。本发明通过rp-hplc,采用主要由离子对试剂和有机改性剂交替使用,组成洗脱溶液进行洗脱,分两步或三步进行纯化,梯度洗脱,可以将待纯化的粗肽中的大部分异构体杂质和其他难以分离的杂质分离、去除;之后采用碳酸氢铵盐溶液作为流动相,进行梯度洗脱,去除难除、微小杂质并有效解决了难洗脱、收率低的问题。实验结果证实,本发明提供的纯化方法所得酸性多肽纯度高、收率高,并且操作简便,有利于实现酸性多肽的规模化制备。
[0015]
有机改性剂的组合搭配方案,以及通过改变有机改性剂的组合浓度,改变有机改性剂与离子对试剂的浓度比,以达到化合物中的某些特定杂质和目标化合物在反相制备达到最大的分离效果。
[0016]
优选地,本发明提供的纯化方法中,洗脱溶剂为nh4cl水溶液和甲醇、乙醇、正丙醇、异丙醇、乙腈中任一种或多种有机改性剂的混合溶液。
[0017]
优选地,本发明提供的纯化方法中,洗脱溶剂为hcooh水溶液和甲醇、乙醇、正丙醇、异丙醇、乙腈中任一种或多种有机改性剂的混合溶液。
[0018]
优选地,本发明提供的纯化方法中,洗脱溶剂为nh4hco3水溶液和甲醇、乙醇、正丙醇、异丙醇、乙腈中任一种或多种有机改性剂的混合溶液。
[0019]
优选地,本发明提供的纯化方法中,洗脱溶剂为teap溶液和甲醇、乙醇、正丙醇、异丙醇、乙腈中任一种或多种有机改性剂的混合溶液。
[0020]
本发明的一些实施例中,分两步进行rp-hplc纯化,洗脱液1为nh4cl水溶液和有机改性剂的混合溶液,洗脱液2为hcooh水溶液和和有机改性剂的混合溶液。
[0021]
本发明的一些实施例中,分三步进行rp-hplc纯化,洗脱液1为teap溶液和有机改性剂的混合溶液,洗脱液2为teap溶液和有机改性剂的混合溶液,洗脱液3为nh4hco3水溶液
和有机改性剂的混合溶液。
[0022]
本发明的一些实施例中,分三步进行rp-hplc纯化,洗脱液1为nh4cl水溶液和有机改性剂的混合溶液,洗脱液2为teap溶液和有机改性剂的混合溶液,洗脱液3为nh4hco3水溶液和有机改性剂的混合溶液。
[0023]
本发明的一些实施例中,分三步进行rp-hplc纯化,洗脱液1为hcooh水溶液和有机改性剂的混合溶液,洗脱液2为nh4cl水溶液和有机改性剂的混合溶液,洗脱液3为nh4hco3水溶液和有机改性剂的混合溶液。
[0024]
本发明的一些实施例中,分三步进行rp-hplc纯化,洗脱液1为nh4cl水溶液和有机改性剂的混合溶液,洗脱液2为hcooh水溶液和有机改性剂的混合溶液,洗脱液3为nh4hco3水溶液和有机改性剂的混合溶液。
[0025]
本发明中所述洗脱液1指分步进行rp-hplc纯化时,第一步纯化时作为流动相的洗脱液;所述洗脱液2指分步进行rp-hplc纯化时,第二步纯化时作为流动相的洗脱液;所述洗脱液3指分步进行rp-hplc纯化时,第三步纯化时作为流动相的洗脱液,以此类推。
[0026]
作为优选,分步进行rp-hplc纯化中,nh4cl作为流动相时,ph为7.0-10.0。
[0027]
作为优选,分步进行rp-hplc纯化中,nh4cl作为流动相时,体积浓度为50mmol/l。
[0028]
作为优选,分步进行rp-hplc纯化中,teap作为流动相时,ph为2.5。
[0029]
作为优选,分步进行rp-hplc纯化中,teap作为流动相时,ph为7.0。
[0030]
作为优选,分步进行rp-hplc纯化中,hcooh作为流动相时,质量浓度为0.1~10%。
[0031]
作为优选,分步进行rp-hplc纯化中,nh4hco3作为流动相时,体积浓度为1~100mmol/l。
[0032]
本发明的一些实施例中,分步进行rp-hplc纯化中,洗脱液1或2的有机改性剂为乙腈、正丙醇、异丙醇中的一种或两种。
[0033]
本发明的一些实施例中,其中分步进行rp-hplc纯化中,洗脱液3的有机改性剂为正丙醇和/或异丙醇。
[0034]
经过一种或两种离子对试剂分离纯化后,再经过最后一种离子对试剂进行分离纯化时采用异丙醇或者正丙醇进行分离纯化,异丙醇或者正丙醇具有较低的毒性,用来替代反相多肽纯化过程中常用毒性较高的乙腈,可以避免药物的临床风险。
[0035]
优选地,本发明提供的纯化方法中,纯化酸性多肽时,使用的离子对试剂的ph大于所分离的酸性多肽化合物pka值至少1个ph单位。
[0036]
采用反相层析的原理,利用反相硅胶分辨率高的特点,结合一种离子对试剂组合的方案,离子对试剂通过不同的ph值的变换,并且所使用的离子对试剂的ph在化合物pka值的1个ph值以上,以保证化合物的结构性质。
[0037]
优选地,本发明提供的纯化方法中,有机改性剂作为流动相时,含量范围为0%~70%。
[0038]
优选地,本发明提供的纯化方法中,含有有机改性剂的流动相梯度范围为20%~60%(60min)。
[0039]
优选地,本发明提供的纯化方法中,反相高效液相色谱系统(rp-hplc)的色谱柱的填料为c4、c6、c8和c18键合硅胶或高分子聚合物。
[0040]
本发明能将全合成、中间过程未进行任何纯化的酸性多肽粗品,尤其是溶解性差
的弱酸性多肽进行分离纯化,得到高纯度终产品,并且保证较高的纯化收率,方法简捷,易操作,节约资源,特别适合于工业化规模制备。
附图说明
[0041]
图1为实施例1粗肽的hplc图谱。
[0042]
图2为实施例1精肽的hplc图谱。
[0043]
图3为实施例16精肽的hplc图谱。
[0044]
图4为实施例17精肽的hplc图谱
具体实施例
[0045]
下面结合具体实施例对本发明的制备方法作进一步详细的说明以便于本领域的技术人员进一步地理解本发明,而不构成对其权利的限制。特别需要指出的是,所有类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明内。
[0046]
实施例1:式ι所示的酸性多肽的制备
[0047]
(1)粗肽的溶解和过滤
[0048]
称取1.03g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.51,然后放置40℃水浴1.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.86%(hplc检测见图,表1)。
[0049]
表1粗肽hplc图谱数据
[0050]
峰号保留时间面积面积%17.4771131141.7227.86386910.1337.99088020.1348.220254670.3958.64359670.0969.106110300.1779.607163950.2589.73484240.13910.01972460.111010.21586410.131110.6351113121.691211.624202360.311312.091969731.471412.557499260.761512.86898641.361613.071397950.601713.529231470.351813.761936921.42
1914.499473826171.862014.854165240.252115.228382270.582215.663235980.362315.8851198721.822416.514927441.412516.993229700.352617.510233410.352718.024777061.182818.713184580.282918.893400060.613019.579283620.433120.35615760.023220.80844560.073321.6284255966.453422.73457830.093523.5251777852.70
[0051]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0052]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0053]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集、并样、旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液。
[0054]
(3)hcooh/乙腈+正丙醇为流动相进行第二次冲洗
[0055]
将上述中间体浓缩液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0056]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样、旋蒸浓缩后冷冻干燥,得到式ι所示的酸性多肽纯品205.3mg,纯度为99.46%,最大单杂为0.19%(hplc检测见图2),图谱数据见表2,总收率为76.3%。
[0057]
表2精肽hplc图谱数据
[0058]
峰号保留时间面积面积%110.4003730.02210.96711440.07311.77125890.15
412.292170770699.46512.91332590.19613.32111560.07713.9428130.05
[0059]
实施例2:式ι所示的酸性多肽的制备
[0060]
(1)粗肽的溶解和过滤
[0061]
称取1.05g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.55,然后放置40℃℃水浴1.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.76%。
[0062]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0063]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0064]
以50mm氯化铵ph7.0为流动相a,以50mm氯化铵ph7.0:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由48%a+52%b(v/v)变为38%a+62%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液。
[0065]
(3)hcooh/乙腈+正丙醇为流动相进行第二次冲洗
[0066]
将上述中间体浓缩液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0067]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为99.02%,最大单杂为0.34%,(hplc检测结果图与图2相似)。
[0068]
实施例3:式ι所示的酸性多肽的制备
[0069]
(1)粗肽的溶解和过滤
[0070]
称取1.02g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.49,然后放置40℃水浴1.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.56%。
[0071]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0072]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0073]
以50mm氯化铵ph10.0为流动相a,以50mm氯化铵ph10.0:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由52%a+48%b(v/v)变为42%a+58%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液。
[0074]
(3)hcooh/乙腈+正丙醇为流动相进行第二次冲洗
[0075]
将上述中间体浓缩液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0076]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为98.88%,最大单杂为0.30%,(hplc检测结果图与图2相似)。
[0077]
实施例4:式ι所示的酸性多肽的制备
[0078]
(1)粗肽的溶解和过滤
[0079]
称取1.00g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.52,然后放置40℃水浴1.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.76%。
[0080]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0081]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0082]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液。
[0083]
(3)hcooh/乙腈+正丙醇为流动相进行第二次冲洗
[0084]
将上述中间体浓缩液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0085]
以5%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由70%a+30%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为98.88%,最大单杂为0.30%,(hplc检测结果图与图2相似)。
[0086]
实施例5:式ι所示的酸性多肽的制备
[0087]
(1)粗肽的溶解和过滤
[0088]
称取1.00g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.85,然后放置40℃℃水浴1.0h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.46%。
[0089]
(2)氯化铵/乙醇为流动相进行第一次冲洗
[0090]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0091]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙醇=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由42%a+58%b(v/v)变为32%a+68%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液。
[0092]
(3)hcooh/乙腈+正丙醇为流动相进行第二次冲洗
[0093]
将上述中间体浓缩液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内
装10μm的c8键合硅胶填料。
[0094]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由72%a+28%b(v/v)变为42%a+58%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为98.59%,最大单杂为0.41%,(hplc检测结果图与图2相似。)。
[0095]
实施例6:式ι所示的酸性多肽的制备
[0096]
(1)粗肽的溶解和过滤
[0097]
称取1.00g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.25,然后放置40℃水浴2.0h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.86%。
[0098]
(2)氯化铵/甲醇为流动相进行第一次冲洗
[0099]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0100]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:甲醇=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由35%a+65%b(v/v)变为25%a+75%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液。
[0101]
(3)hcooh/乙腈+正丙醇为流动相进行第二次冲洗
[0102]
将上述中间体浓缩液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0103]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为98.88%,最大单杂为0.30%,(hplc检测结果图与图2相似)。
[0104]
实施例7:式ι所示的酸性多肽的制备
[0105]
(1)粗肽的溶解和过滤
[0106]
称取1.02g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.5
±
0.5,然后放置40℃水浴1.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.56%。
[0107]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0108]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c18键合硅胶填料。
[0109]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液。
[0110]
(3)nh4hco3/乙腈为流动相进行第二次冲洗
[0111]
将上述中间体浓缩液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0112]
以10mm碳酸氢铵为流动相a,以乙腈:纯化水=800:200(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由60%a+40%b(v/v)变为50%a+50%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,得到式ι所示的酸性多肽纯品198.6mg,检测冻干后样品纯度为98.49%,最大单杂为0.63%,(hplc检测结果图与图2相似)。
[0113]
实施例8:式ι所示的酸性多肽的制备
[0114]
(1)粗肽的溶解和过滤
[0115]
称取1.03g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.95,然后放置40℃水浴1.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.86%。
[0116]
(2)1%teap ph7.0/乙腈为流动相进行第一次冲洗
[0117]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0118]
以1%teap ph7.0为流动相a,以1%teap ph7.0:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由45%a+55%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0119]
(3)1%teap ph2.5/正丙醇为流动相进行第二次冲洗
[0120]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0121]
以1%teap ph2.5为流动相a,以1%teap ph2.5:正丙醇=300:700(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由62%a+38%b(v/v)变为52%a+48%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液2。
[0122]
(4)nh4hco3/乙腈为流动相进行第三次冲洗
[0123]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0124]
以10mm碳酸氢铵为流动相a,以乙腈:纯化水=800:200(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由60%a+40%b(v/v)变为50%a+50%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,得到式ι所示的酸性多肽纯品178.5mg,检测冻干后样品纯度为98.56%,最大单杂为0.49%(hplc检测结果图与图2相似)。
[0125]
实施例9:式ι所示的酸性多肽的制备
[0126]
(1)粗肽的溶解和过滤
[0127]
称取1.04g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超
声溶解,加入三乙胺至完全溶清,再用tfa调ph7.38,然后放置40℃水浴1.0h,使用0.45μm有机滤膜过滤。检测粗肽溶液,粗肽hplc纯度70.86%。
[0128]
(2)1%teap ph2.5/正丙醇为流动相进行第一次冲洗
[0129]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0130]
以1%teap ph2.5为流动相a,以1%teap ph2.5:正丙醇=300:700(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由62%a+38%b(v/v)变为52%a+48%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0131]
(3)1%teap ph7.0/乙腈为流动相进行第二次冲洗
[0132]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0133]
以1%teap ph7.0为流动相a,以1%teap ph7.0:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由45%a+55%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽的制备中间体浓缩液2。
[0134]
(4)nh4hco3/乙腈为流动相进行第三次冲洗
[0135]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0136]
以10mm碳酸氢铵为流动相a,以乙腈:纯化水=800:200(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由60%a+40%b(v/v)变为50%a+50%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为98.56%,最大单杂为0.49%(hplc检测结果图与图2相似)。
[0137]
实施例10:式ι所示的酸性多肽的制备
[0138]
(1)粗肽的溶解和过滤
[0139]
称取1.03g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.68,然后放置40℃水浴1.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度71.66%。
[0140]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0141]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0142]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0143]
(3)1%teap ph7.0/乙腈为流动相进行第二次冲洗
[0144]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0145]
以1%teap ph7.0为流动相a,以1%teap ph7.0:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由45%a+55%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液2。
[0146]
(4)nh4hco3/乙腈为流动相进行第三次冲洗
[0147]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0148]
以10mm碳酸氢铵为流动相a,以乙腈:纯化水=800:200(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由60%a+40%b(v/v)变为50%a+50%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为98.85%,最大单杂为0.34%(hplc检测结果图与图2相似)。
[0149]
实施例11:式ι所示的酸性多肽的制备
[0150]
(1)粗肽的溶解和过滤
[0151]
称取1.01g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.5
±
1.0,然后放置40℃
±
10℃水浴1.5
±
0.5h,使用0.45μm有机滤膜过滤,检测粗肽溶液,粗肽hplc纯度72.46%。
[0152]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0153]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0154]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0155]
(3)1%teap ph2.5/正丙醇为流动相进行第二次冲洗
[0156]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0157]
以1%teap ph2.5为流动相a,以1%teap ph2.5:正丙醇=300:700(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由62%a+38%b(v/v)变为52%a+48%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液2。
[0158]
(4)nh4hco3/乙腈为流动相进行第三次冲洗
[0159]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0160]
以10mm碳酸氢铵为流动相a,以乙腈:纯化水=800:200(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由60%a+40%b(v/v)变为50%a+50%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,并样旋蒸浓缩后冷冻干燥,检测冻干后样品纯度为98.97%,最大单杂为0.34%(hplc检测结果图与图2相似)。
[0161]
实施例12:式ι所示的酸性多肽的制备
[0162]
(1)粗肽的溶解和过滤
[0163]
称取1.03g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.58,然后放置45℃水浴1.5h,使用0.45μm有机滤膜过滤。检测粗肽溶液,粗肽hplc纯度71.16%。
[0164]
(2)hcooh/乙腈为流动相进行第一次冲洗
[0165]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0166]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0167]
(3)氯化铵/乙腈为流动相进行第二次冲洗
[0168]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0169]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液2。
[0170]
(4)nh4hco3/乙腈为流动相进行第三次冲洗
[0171]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0172]
以10mm碳酸氢铵为流动相a,以乙腈:纯化水=800:200(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由60%a+40%b(v/v)变为50%a+50%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,得到式ι所示的酸性多肽纯品198.7mg,检测冻干后样品纯度为99.14%,最大单杂为0.31%(hplc检测结果图与图2相似)。
[0173]
实施例13:式ι所示的酸性多肽的制备
[0174]
(1)粗肽的溶解和过滤
[0175]
称取1.02g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.68,然后放置45℃水浴1.5h,使用0.45μm有机滤膜过滤。检测粗肽溶液,粗肽hplc纯度71.06%。
[0176]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0177]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0178]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液
相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0179]
(3)hcooh/乙腈为流动相进行第二次冲洗
[0180]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0181]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液2。
[0182]
(4)nh4hco3/乙腈为流动相进行第三次冲洗
[0183]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0184]
以10mm碳酸氢铵为流动相a,以乙腈:纯化水=800:200(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由60%a+40%b(v/v)变为50%a+50%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,得到式ι所示的酸性多肽纯品203.6mg,检测冻干后样品纯度为99.62%,最大单杂为0.13%(hplc检测结果图与图2相似)。
[0185]
实施例14:式ι所示的酸性多肽的制备
[0186]
(1)粗肽的溶解和过滤
[0187]
称取1.01g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.55,然后放置45℃水浴1.5h,使用0.45μm有机滤膜过滤。检测粗肽溶液,粗肽hplc纯度72.06%。
[0188]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0189]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0190]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0191]
(3)hcooh/乙腈为流动相进行第二次冲洗
[0192]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0193]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液2。
[0194]
(4)nh4hco3/正丙醇为流动相进行第三次冲洗
[0195]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0196]
以10mm碳酸氢铵为流动相a,以纯化水:正丙醇=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由61%a+39%b(v/v)变为41%a+59%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,得到式ι所示的酸性多肽纯品209.9mg,检测冻干后样品纯度为99.55%,最大单杂为0.12%(hplc检测结果图与图2相似)。
[0197]
实施例15:式ι所示的酸性多肽的制备
[0198]
(1)粗肽的溶解和过滤
[0199]
称取1.05g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.63,然后放置45℃水浴1.5h,使用0.45μm有机滤膜过滤。检测粗肽溶液,粗肽hplc纯度70.86%。
[0200]
(2)氯化铵/乙腈为流动相进行第一次冲洗
[0201]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0202]
以50mm氯化铵ph8.5为流动相a,以50mm氯化铵ph8.5:乙腈=300:700作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由50%a+50%b(v/v)变为40%a+60%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液1。
[0203]
(3)hcooh/乙腈+和正丙醇为流动相进行第二次冲洗
[0204]
将上述中间体浓缩液1通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0205]
以0.1%hcooh为流动相a,以正丙醇:乙腈=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由67%a+33%b(v/v)变为37%a+63%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽中间体浓缩液2。
[0206]
(4)nh4hco3/异丙醇为流动相进行第三次冲洗
[0207]
将上述中间体浓缩液2通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0208]
以10mm碳酸氢铵为流动相a,以纯化水:异丙醇=1:1(v/v)作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由55%a+45%b(v/v)变为35%a+65%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集纯度>98%的馏分合并,并样旋蒸浓缩后冷冻干燥,得到式ι所示的酸性多肽纯品208.8mg,检测冻干后样品纯度为99.62%,最大单杂为0.14%(hplc检测结果图与图2相似)。
[0209]
实施例16:式ι所示的酸性多肽的制备
[0210]
(1)粗肽的溶解和过滤
[0211]
称取1.02g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.76,然后放置45℃水浴1.5h,使用0.45μm有机滤膜过滤。检测粗肽溶液,粗肽hplc纯度72.06%。
[0212]
(2)0.1%tfa/乙腈为流动相进行冲洗
[0213]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0214]
以0.1%tfa为流动相a,以乙腈作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由63%a+37%b(v/v)变为43%a+57%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽纯品,检测纯度为96.39,最大单杂为2.65%(hplc检测见图3,图谱数据见表3)。
[0215]
表3精肽hplc图谱数据
[0216][0217][0218]
实施例17:式ι所示的酸性多肽的制备
[0219]
(1)粗肽的溶解和过滤
[0220]
称取1.05g粗肽,碾碎后加入200ml溶剂(乙腈:纯化水=20:80的体积比例混合)超声溶解,加入三乙胺至完全溶清,再用tfa调ph7.59,然后放置45℃水浴1.5h,使用0.45μm有机滤膜过滤。检测粗肽溶液,粗肽hplc纯度72.86%。
[0221]
(2)0.1%磷酸/乙腈为流动相进行冲洗
[0222]
将上述过滤后的粗肽溶液通过进样泵加载到色谱柱上,色谱柱的规格为30*250mm,内装10μm的c8键合硅胶填料。
[0223]
以0.1%磷酸为流动相a,以乙腈作为流动相b,洗脱流速为25ml/min,梯度洗脱洗脱方式为:60min内,梯度由65%a+35%b(v/v)变为45%a+55%b(v/v),启动流动相洗脱,采集图谱,观察吸收值的变化,收集主峰并用分析液相检测纯度,收集并样旋蒸浓缩去除乙腈后,得到式ι所示的酸性多肽纯品,检测纯度为93.05%,最大单杂为2.60%(hplc检测见图4,图谱数据见表4),。
[0224]
表4精肽hplc图谱数据
[0225]
峰号保留时间面积面积%17.40011080.0827.81912370.0938.87513950.1049.10812670.0959.40430100.2269.71531430.23710.30617940.13811.68968910.49
912.44062160.451012.792100260.721113.198118020.851214.074129692993.051314.871362742.601416.26889330.641518.72622530.161621.18416020.11
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询




tips