
 商标分类
商标分类  商标转让
商标转让 
肼硼烷的合成方法与流程
 2021-01-31 04:01:01|
2021-01-31 04:01:01| 278|
278| 起点商标网
起点商标网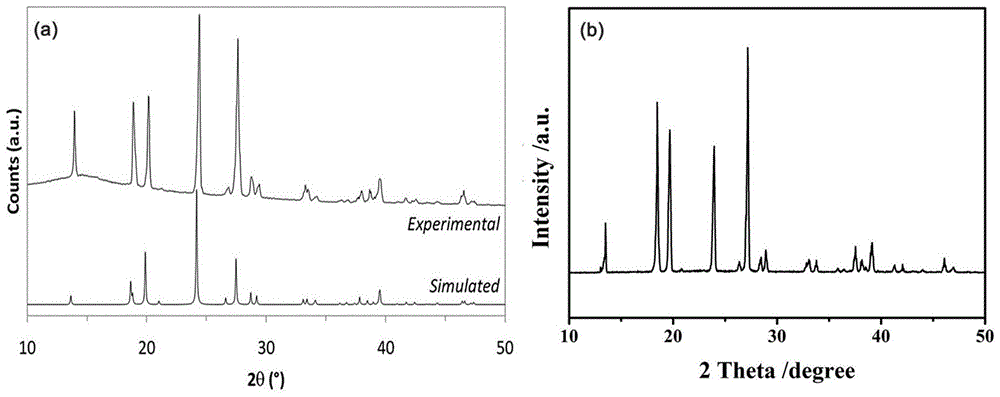
本发明涉及有机物合成技术领域,具体涉及有机物肼硼烷的合成方法。
背景技术:
氢气是一种有前途的能源载体,它将促进从化石燃料向可再生能源的过渡,向可持续能源的未来发展。在越来越受到关注的同时,找到有效的储氢材料是基于h2经济的燃料电池最困难的挑战之一。在各种类型的储氢材料中,化学储氢被认为是安全有效储氢的最有前途的方法之一。一些硼氢化物,硼烷及其衍生物等由于具有高储氢密度、相对稳定、安全的、易加工性而备受关注。
肼硼烷(hb,n2h4bh3)的含氢量高达15.4wt%,在室温下是物理化学性质稳定安全的固体,是一种极具潜力的化学储氢材料。肼硼烷可以通过热解、醇解和水解产氢。特别是,通过水解其硼烷基和选择性裂解肼基实现完全产氢后,其对应的n2h4bh3-3h2o系统的有效理论质量储氢容量达10wt%,远高于已知的氢源系统nabh4-4h2o(7.3wt%),nh3bh3-4h2o(5.9wt%)和n2h4·h2o(8.0wt%)。理论上,1摩尔hb可以通过bh3基团的水解释放5摩尔h2,方程式是:[n2h4bh3(s)+3h2o→b(oh)3(l)+n2(g)+5h2(g)]。
然而,目前市面上并无商业化的肼硼烷出现。经报道,physicalchemistrychemicalphysics,2012,14,1768-1777公开了肼硼烷的合成方法,该方法合成的肼硼烷的纯度为99.6%,收率仅为81.6%。而且,该方法需要在30℃加热条件下合成3天才可以完成。因此,设计出操作简便的高收率和高纯度的肼硼烷的合成路线,将从经济角度为实现肼硼烷商业化的宏伟目标作出很大的贡献。
技术实现要素:
本发明为了克服上述技术问题,提供了一种收率能达到90%和纯度为99.9%的肼硼烷的合成路线,在室温条件下进行,并且只需要反应12~48h即可完成。
解决上述技术问题的技术方案如下:
肼硼烷的合成方法,包括以下步骤:
1)室温下,将肼盐和还原剂按照一定比例放入反应器中,再加入适量的溶剂;
2)向反应器内通入惰性气体,室温下搅拌反应12~48h;
3)将步骤2)的反应器内的混合物进行离心分离,并将上清液进行真空干燥得白色晶体即为肼硼烷粗产品;
4)将步骤3)得到的白色晶体用正戊烷离心洗涤2-3次,洗涤后在一定温度下真空干燥,干燥后的白色固体即为肼硼烷。
进一步地说,步骤1)所述的肼盐为肼半硫酸盐(n2h4·1/2h2so4),盐酸肼(n2h4·hcl),四肼氯化镁(mgcl2·4n2h4)或肼(n2h4)中的任意一种。
优选为肼半硫酸盐(n2h4·1/2h2so4)。
进一步地说,步骤1)所述的还原剂为硼氢化钠(nabh4),硼氢化钾(kbh4),硼氢化锂(libh4),硼氢化铝(al(bh4)3)或硼烷(b2h6)中的任意一种。
进一步地说,步骤1)所述的溶剂为1,4二氧六环(c4h8o2),四氢呋喃(thf),苯(c6h6),庚烷(c7h16),己烷(c6h14),肼(n2h4)中的任意一种。
优选溶剂为1,4二氧六环(c4h8o2)。
进一步地说,步骤1)所述的肼盐和还原剂按照摩尔比为1:1~2放入反应器中,所述的溶剂的加入体积量ml至少是还原剂质量g的8倍,所述的溶剂的加入体积量ml至少是肼盐质量g的3倍。
进一步地说,当溶剂为苯(c6h6),庚烷(c7h16),己烷(c6h14),肼(n2h4)时,步骤3)是将步骤2)的反应器的混合物用thf进行萃取,然后再在n2氛围中蒸发掉thf得到肼硼烷粗产品。
进一步地说,步骤2)所述的惰性气体为氩气。
更进一步地说,步骤3)和步骤4)所述的真空干燥的温度均为303~313k。
本发明的有益效果是:
该发明中通过简便的方法合成了稳定的白色肼硼烷晶体,收率能达到90%,纯度可达到99.9%,在室温条件下进行,并且只需要反应12~48h即可完成。本发明方法合成出的肼硼烷具有收益普遍好,含氢量高等特点,可以作为一种非常有前景的储氢材料。
附图说明
下面结合附图和具体实施方式对本发明作进一步详细的说明。
图1a为背景技术中所述的physicalchemistrychemicalphysics,2012,14,1768-1777公开的方法合成的肼硼烷的x射线衍射谱图(xrd);
图1b为本发明实施例1合成的肼硼烷的x射线衍射谱图(xrd);
图2和图3为本发明实施例1合成的肼硼烷的液体核磁共振(nmr),其中图2为11bnmr,图3为1hnmr。
图4为本发明实施例1合成的肼硼烷的傅里叶变换红外谱图(ftir)。
具体实施方式
实施例1:
将21.67g(0.26mol)n2h4.1/2h2so4和10.0g(0.26mol)nabh4放入双颈烧瓶,再倒入80ml1,4二氧六环,接着通入氩气,室温下搅拌48h。搅拌完成后,将反应后的产物离心,将分离出的上清液进行313k的真空干燥。干燥完成后,将干燥后的固体产物用正戊烷洗涤两次,再进行313k的真空干燥。即可获得目标产物肼硼烷。
分别采用x射线衍射谱图(xrd)、液体核磁共振(nmr)和傅里叶变换红外谱图(ftir)对实施例1合成的产物肼硼烷进行晶型结构分析、化学结构和纯度分析、化学结构分析。
1)通过x射线衍射谱图(xrd)进行晶型结构比对分析
附图1a是背景技术中记载的physicalchemistrychemicalphysics,2012,14,1768-1777公开的方法合成的肼硼烷的x射线衍射谱图(xrd);附图1b是本实施例合成的肼硼烷的x射线衍射谱图(xrd),经过比对发现,本发明的峰的位置、晶面间距、强度和半峰宽,与moury等人利用正交晶系pbcn(60)空间群进行理论计算的理论值一致,定性说明本发明方法成功合成了高纯度的肼硼烷。
2)通过液体核磁共振(nmr)分析肼硼烷的化学结构和纯度
将实施例1所制备出的白色肼硼烷晶体取40mg溶于600ml的cd3cn,随后将所得溶液放进核磁管进行核磁共振检测。其液体硼核磁共振图谱(11bnmr)在-18.5到-15ppm有一个特征信号,在11b{1h}图谱中出现一个四重峰(1:3:31)归于bh3基团(1jbh94±1hz),具体见图2。n2h4基团可以由1hnmr图谱证明,具体见图3,图中位于3.44ppm和5.5ppm的两个单峰,分别归因于nh2-n的2h和nh2-b的2h。由于11b和1h之间的杂核耦合作用,bh3基团还可以通过位于1到1.7ppm处的四重峰(1:1.1:1.1:1)来确定。此外,一些小信号(三个位于1.10-1.6ppm的可见峰和一个四重叠峰)属于11b和1h之间的杂核耦合。通过计算,该肼硼烷的纯度高于99.9%。
3)通过傅里叶变换红外谱图(ftir)进行化学结构分析
将实施例1所制备出的白色肼硼烷晶体进行傅里叶变换红外谱图分析,具体见图4。图4显示了其频带可以按以下方式分配:(i)在2600-3600cm-1范围内的n-h拉伸区域中具有不同强度的多个频带;(ii)b-h伸展区有一条大条带(1975-2600cm-1);(iii)b-h伸展区观察到一条小条带(1715和1975cm-1),表明bh3的h与其他元素之间发生了强相互作用;(iv)在n-h非对称弯曲区域(1600和1630cm-1)中形成了2条非常强烈的尖锐带。(v)在n-h的对称弯曲区域(1435和1335cm-1)有2条尖锐的强带;(vi)b-h弯曲区域(1100-1300cm-1)中也有条带;(vii)在n-h区有2条锐带(1045和980cm-1);(viii)bn-n不对称拉伸区域和n-n对称拉伸区域中的一条强带(910cm-1);(ix)在bn-n拉伸区域和bn-n对称拉伸区域(750cm-1)的中等频带。这些数据符合最初报道的波数和属性,因此支持hb的分子结构。
实施例2:
将13.6g(0.2mol)n2h4·hcl和4.4g(0.2mol)libh4放入反应器内,再倒入250mlthf,接着通入氮气,并在室温下搅拌48h。搅拌完成后,将反应后的产物离心,将分离出的上清液进行303k的真空干燥。干燥完成后,将干燥后的固体产物用正戊烷洗涤两次,再进行313k的真空干燥。即可获得目标产物肼硼烷。
实施例3:
在室温下,将1.8g(0.008mol)mgcl2·4n2h4与0.6g(0.016mol)nabh4混合。溶于50mlc7h16溶液。并将所得混合物连续搅拌直至不再放出气体(12h)。用四氢呋喃萃取固体残留物。在氮气下蒸发四氢呋喃后,用正戊烷洗涤三次,再进行310k的真空干燥。即可获得目标产物肼硼烷。
实施例4
在室温下,将1.38g(0.02mol)n2h4·hcl,0.84g(0.03mol)b2h6和100mln2h4一起加入双颈烧瓶中一起搅拌。在此过程中通入ar,直至制氢停止(24h)。反应完成后,离心分离出固体,并将得到的清液进行真空干燥,得到白色固体。最后再用正戊烷进行洗涤,真空干燥后即可获得目标产物。
实施例5
通过0.32g(0.1mol)n2h4和6g(0.15mol)kbh4在500mlc6h14中反应来制备肼硼烷。在室温下和氮气氛围中搅拌1天。最后,将反应混合物离心分离并干燥。即可获得目标产物。
实施例6
在室温下,将0.68g(0.01mol)n2h4·hcl和0.72g(0.01mol)al(bh4)3加入到50ml干燥的苯中。同时通入惰性气体,搅拌反应混合物,注意到放出氢气,并且反应容器中的固体转化为胶状物质。当氢气停止逸出时(2天),倾析出透明的烃液体,并用四氢呋喃萃取固体残留物。在氮气下蒸发四氢呋喃后,即可获得目标产物。
以上所述,仅是本发明的较佳实施例,并非对本发明做任何形式上的限制,凡是依据本发明的技术实质上对以上实施例所作的任何简单修改、等同变化,均落入本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



