
 商标分类
商标分类  商标转让
商标转让 
六方氮化硼膜的合成和转移方法与流程
 2021-01-31 02:01:51|
2021-01-31 02:01:51| 395|
395| 起点商标网
起点商标网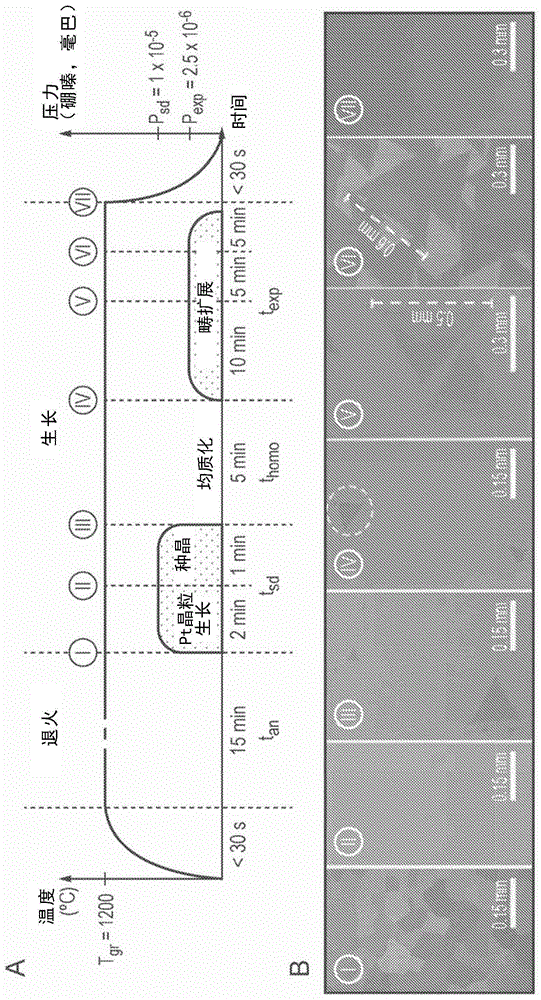
技术领域和
背景技术:
本公开涉及合成高质量六方氮化硼的方法。这对于(但不限于)制造包含多种二维材料(2dm)的异质结构特别有用。
近年来,由于在曼彻斯特大学的andregeim和konstantinnovoselov于2004年分离石墨烯(由此他们于2010年获得诺贝尔物理学奖)后2dm的令人印象深刻的独特固有性质,所以对2dm的潜力感兴趣。
在分离石墨烯后,还分离了许多其他2dm。特别令人感兴趣的是六方氮化硼(h-bn),其晶格参数和层间间距与石墨烯类似,但与作为零间隙半导体的石墨烯不同,h-bn是绝缘体。这使得h-bn成为理想的石墨烯基底,由于基底表面的起伏以及石墨烯与基底的相互作用,石墨烯通常在典型的基底上显示出极大的性能下降。因此,对制造包含多层石墨烯和h-bn的“异质结构”设备感兴趣。
迄今为止,由于难以生产可以将其从生长转移到器件基底的大晶片规模面积的石墨烯和h-bn,因此仅制造了小面积的石墨烯-h-bn异质结构器件。
最初,石墨烯和h-bn都使用物理剥离技术制造,而物理剥离技术虽然生产高质量的材料但其规模通常限制在几十微米。已经开发了其他大规模制造技术,主要是化学气相沉积(cvd),这些制造技术能够在晶片规模(数十厘米)上制造石墨烯和h-bn。
尽管已显示cvd能够制造晶片规模石墨烯和h-bn,但是证明在不显著降低材料固有性质的情况下,很难将石墨烯和h-bn转移到器件基底。为了解决在转移cvd生长的石墨烯方面的困难,已经进行最新的尝试,但是迄今为止,还没有论证出在保持高水平固有性质的同时生长和转移cvd生长的h-bn的实用方法。
如上所述,本公开的至少某些实施方式解决了这些问题中的一个或更多个。
技术实现要素:
在所附的权利要求书中提出具体的方面和实施方式。
从一个角度来看,可以提供一种通过化学气相沉积在基底上生产六方氮化硼的方法,所述方法包括:(a)在第一温度下加热所述基底长达第一时间的步骤;(b)在第二温度下在前体的第一分压下将所述基底暴露到包含硼的前体和包含氮的前体长达第二时间的步骤,其中将单一前体用作包含硼的前体和包含氮的前体或将不同前体用作包含硼的前体和包含氮的前体;(c)在不具有前体的情况下在第三温度下加热所述基底长达第三时间的步骤;和(d)在第四温度下在前体的第二分压下将所述基底暴露到所述前体长达第四时间的步骤。
可以认为本方法分为四个主要步骤:(a)退火步骤;(b)重结晶/种晶步骤;(c)均质化步骤;和(d)畴扩展步骤。步骤(a)有助于从基底上清除碎屑,减少基底上的表面杂质并促进基底的初始重结晶。骤(b)提供了多个h-bn晶畴的初始成核。步骤(b)还可以促进基底材料的进一步重结晶,从而导致可以在其上形成h-bn的基底中更大的均匀畴。步骤(c)起到收缩在步骤(b)期间形成的h-bn晶畴的作用,从而减少现存晶畴的数量。步骤(d)起到扩展剩余晶畴同时使进一步成核位点的形成最小化的作用。
因此,在步骤(b)中的初始成核和步骤(d)中的畴扩展之间的分裂允许少量的大面积h-bn晶畴生长。
在一些实施例中,第二分压低于第一分压。因此,可以提供在步骤(b)中促进成核并在步骤(d)中抑制成核,同时仍然允许畴扩展的条件。
在一些实施例中,第一分压是1x10-6毫巴至1x10-2毫巴且第二分压是1x10-7毫巴至1x10-2毫巴。
在其他实施例中,第一分压是5x10-6毫巴至1.5x10-5毫巴且第二分压是1x10-6毫巴至4x10-6毫巴。在优选的实施例中,第一分压是9x10-6毫巴至1.1x10-5毫巴且第二分压是2x10-6毫巴至3x10-6毫巴。
在一些实施例中,第一分压可以是5x10-6毫巴、6x10-6毫巴、7x10-6毫巴、8x10-6毫巴、9x10-6毫巴、1.0x10-5毫巴、1.1x10-5毫巴、1.2x10-5毫巴、1.3x10-5毫巴、1.4x10-5毫巴和1.5x10-5毫巴中的任一个或介于这些数值之间的任何子范围。在一些实施例中,第二分压可以是1x10-6毫巴、1.5x10-6毫巴、2x10-6毫巴、2.5x10-6毫巴、3x10-6毫巴、3.5x10-6毫巴和4x10-6毫巴中的任一个或介于这些数值之间的任何子范围。因此,可以提供在步骤(b)中促进成核并在步骤(d)中抑制成核,同时仍然允许畴扩展的条件。
在一些实施例中,单一前体用作包含硼的前体和包含氮的前体,并且其中该前体是硼嗪、氨硼烷和三氯硼嗪中的一种。因此,可以提供单一前体,其既充当硼前体又充当氮前体,因此简化了向生长室的前体递送,因为仅需要递送单一气体并且需要控制单一相关联的压力。
在一些实施例中,包含硼的前体是硼酸三异丙酯、三苯基硼烷、三氯化硼、乙硼烷和十硼烷中的一种;包含氮的前体是氨气(ammonia)和氮气(nitrogen)中的一种。因此,可以分别控制可用硼和氮的量,从而允许进一步细化生长和重结晶条件。
在一些实施例中,基底是铂或铂合金。铂是h-bn生长的良好催化剂并且在生长后对h-bn的粘附性较弱。因此,铂的使用促进生长后h-bn向器件基底的转移。术语器件基底(devicesubstrate)的使用例如是指期望在生长之后在其上沉积h-bn的任何靶基底。可以用作生长h-bn的催化剂的具有弱粘附性的催化剂的其他实例是锗、铜、银、金、铱和包括一种或多种这些物类的合金。使用弱粘附性的材料作为基底促进基底的重复使用,因为可以以无损方式(例如剥离)从基底中去除h-bn,从而可以重新使用基底。因此,可以通过重复使用来抵消使用铂的高成本。
铂的使用允许在cvd生长过程期间选择参数时访问广泛的参数空间,因为铂在高温下是化学惰性的且因此可以使用高温。这种化学惰性进一步使对表面处理(例如氧化物去除)的需求最小化。此外,在步骤(b)中,铂基底可以经历重结晶,这通过最好地理解是将硼从硼前体溶解到铂基底中来增加铂晶畴的尺寸。溶解的硼起脱氧剂的作用,该脱氧剂除去存在于晶界的、抑制晶粒生长的杂质。铂晶畴尺寸的增加继而促进h-bn的大型单晶畴的生长。在基底由纯金属或合金形式的其他金属形成的其他实施例中,硼的脱氧剂作用还起到去除存在于晶界的、抑制晶粒生长的杂质的作用,因此也促进基底晶畴的尺寸增加。
在一些实施例中,基底是铂箔。因此,可以容易地制备铂基底并且易于使用。在其他实施例中,可以使用沉积的铂薄膜或块铂。
在一些实施例中,基底由单晶铂形成。因此,铂基底的畴尺寸增加,从而促进h-bn的大单晶畴的生长。
在一些实施例中,基底最初由多晶铂形成,并且步骤(b)引起铂基底从多晶形式重结晶为单晶形式。因此,铂基底的晶畴尺寸增加,从而促进h-bn的大单晶畴的生长。
在一些实施例中,第一温度是900℃至1400℃。在其他实施例中,第一温度是1170℃至1250℃。在优选的实施例中,第一温度是1180℃至1220℃。在一些实施例中,第一温度可以是1150℃、1160℃、1170℃、1180℃、1190℃、1200℃、1210℃、1220℃、1230℃、1240℃、1250℃、1300℃和1350℃中的任一个或这些数值之间的任何子范围。
因此,为基底提供有足够的热量以清除碎屑,减少基底上的表面杂质并促进基底的初始重结晶而不会引起基底熔化。
在一些实施例中,第二温度是900℃至1400℃。在其他实施例中,第二温度是1170℃至1250℃。在优选的实施例中,第二温度是1180℃至1220℃。在一些实施例中,第二温度可以是1150℃、1160℃、1170℃、1180℃、1190℃、1200℃、1210℃、1220℃、1230℃、1240℃、1250℃、1300℃和1350℃中的任一个或这些数值之间的任何子范围。
因此,成核速率保持为低的非零速率。此外,该温度足以允许在合适的基底例如铂中进一步重结晶。
在一些实施例中,第三温度是900℃至1400℃。在其他实施例中,第三温度是1170℃至1250℃。在优选的实施例中,第三温度是1180℃至1220℃。在一些实施例中,第三温度可以是1150℃、1160℃、1170℃、1180℃、1190℃、1200℃、1210℃、1220℃、1230℃、1240℃、1250℃、1300℃和1350℃中的任一个或这些数值之间的任何子范围。
因此,温度足够高以允许在步骤(b)中形成的h-bn晶体的至少一部分解离,同时又足够低以至于并非所有这样的晶体都解离。
在一些实施例中,第四温度是900℃至1400℃。在其他实施例中,第四温度是1170℃至1250℃。在优选的实施例中,第四温度是1180℃至1220℃。在一些实施例中,第四温度可以是1150℃、1160℃、1170℃、1180℃、1190℃、1200℃、1210℃、1220℃、1230℃、1240℃、1250℃、1300℃和1350℃中的任一个或这些数值之间的任何子范围。
因此,温度足以允许存在的h-bn晶畴的畴扩展,同时抑制进一步成核位点的形成。
在一些实施例中,第一、第二、第三和第四温度基本上相同。因此,可以将cvd生长室保持在恒定温度,从而简化控制并减少与增加和降低室的温度相关的能量。
在一些实施例中,第一时间是至少5分钟。在一些实施例中,第一时间可以等于或大于10秒、5分钟、10分钟、15分钟、20分钟、25分钟和30分钟中的任一个。在优选的实施例中,第一时间是至少10分钟。因此,时间足以允许清除碎屑并减少基底上的表面杂质。
在一些实施例中,第二时间是1分钟至10分钟。在其他实施例中,第二时间是2至6分钟。在优选的实施例中,第二时间是3至4分钟。在一些实施例中,第二时间可以是1分钟、2分钟、3分钟、4分钟、5分钟、6分钟、7分钟、8分钟、9分钟、10分钟或30分钟中的任一个或这些数值之间的任何子范围。
因此,该时间足够长以允许形成初始成核位点,但时间不足够长使得没有显著连续轮次形成进一步的成核位点。因此,使成核位点的数目最小化,这有助于形成大的h-bn晶畴。
在一些实施例中,第三时间是1分钟至30分钟。在其他实施例中,第三时间是2分钟至10分钟。在优选的实施例中,第三时间是4分钟至6分钟。在一些实施例中,第三时间可以是1分钟、2分钟、3分钟、4分钟、5分钟、6分钟、7分钟、8分钟、9分钟、10分钟或30分钟中的任一个或这些数值之间的任何子范围。
因此,该时间足以允许h-bn晶畴的部分解离,同时仍然留下足够数量的晶畴以允许在步骤(d)中的h-bn期间足够的生长。以这种方式,应当理解,小晶畴将完全解离,并且在步骤(d)之后,存在相对少数量的大面积h-bn单晶畴。
在一些实施例中,第四时间是5分钟至60分钟。在其他实施例中,第四时间是5分钟至20分钟。在优选的实施例中,第四时间是8分钟至12分钟。在一些实施例中,第四时间可以是5分钟、6分钟、7分钟、8分钟、9分钟、10分钟、11分钟、12分钟、13分钟、14分钟、15分钟、16分钟、17分钟、18分钟、19分钟、20分钟、30分钟和60分钟中的任一个或这些数值之间的任何子范围。
因此,该时间足以允许现存的h-bn晶畴的畴扩展,同时抑制其他成核位点的形成。
从一个角度来看,可以提供一种将通过前述方法中的任何一种生产的六方氮化硼从第一基底转移到第二基底的方法,所述方法包括:(e)将载体材料施加到六方氮化硼,所述载体材料对六方氮化硼的粘附性高于六方氮化硼对第一基底的粘附性,使得六方氮化硼粘附到载体材料;(f)从第一基底除去附着有六方氮化硼的载体材料;(g)将粘附有六方氮化硼的载体材料施加到第二基底;和(h)去除载体材料。
因此,本方法提供了一种方法,通过该方法可以将h-bn从生长基底转移到器件基底。该方法使对生长基底的损害最小化,从而允许生长基底的重复使用,这对于高成本的催化剂如铂尤其重要。此外,该方法使生长的h-bn的污染最小化,从而保持h-bn的所需高质量固有性质。从一个角度来看,本方法可以被认为是一种基于剥离的方法,该方法使用由载体材料制成的“压模(stamp)”。例如,术语“压模(stamp)”可以被认为是指有助于处理附着/粘附到压模的材料的材料主体。在一些实施例中,将载体材料滴铸到生长的h-bn,随后将载体材料与h-bn一起剥离,然后将具有h-bn的载体材料沉积在新的基底上且最后去除载体材料。
本方法与例如依赖溶解生长基底(例如湿转移)的现有技术相反。如本发明人所确定的,这污染生长的h-bn,并且有必要为随后的生长运行提供新的生长基底。其他现有技术,例如电化学分层,尽管避免了溶解生长基底的需要,但仍然对生长基底和生长的h-bn都表现出实质性的污染。
在一些实施例中,载体材料是lor(抗剥离剂(lift-offresist))、pmma(聚甲基丙烯酸甲酯)、ppc(聚碳酸亚丙酯)、pvb(聚乙烯醇缩丁醛)、cab(醋酸纤维素丁酸酯)、pvp(聚乙烯吡咯烷酮)、pc(聚碳酸酯)或pva(聚乙烯醇)。因此,提供了载体材料,其对h-bn的粘附性可以高于h-bn对生长基底的粘附性,并且不会对生长基底或h-bn造成损害。
在一些实施例中,步骤(e)-(h)被重复多次以在第二基底上建立多层六方氮化硼。因此,可以容易地制造多层h-bn。在一些实施例中,可以容易地制造精确数量的h-bn层。在一些实施例中,由于h-bn层与第二h-bn层的粘附性可以比第二h-bn层对其基底的粘附性强,所以载体层与附着的h-bn层一起可以共同用作“压模”以拾取其他h-bn层。
类似地,可以使用载体层与附着的h-bn层的“压模”来制造包含h-bn和其他二维材料的多个混合层的异质结构,所述其他二维材料包括但不限于石墨烯、石墨烯衍生物和过渡金属二卤化物。可以制造这种异质结构,因为h-bn层对二维材料层的粘附性可能强于二维材料层对二维材料层的生长基底的粘附性。
可以使用载体层与附着的h-bn层的“压模”来制造将h-bn用作覆盖层的常规结构。这包括任何材料的金属、半导体和绝缘层的转移,只要这些层对h-bn的粘附性大于这些层对它们各自的基底的粘附性。
在一些实施例中,第二基底是硅、二氧化硅、氧化铝、蓝宝石、锗、砷化镓(gaas)、硅和锗的合金以及磷化铟中的一种。因此,可以将h-bn转移到适合于器件制造的基底。应当理解,上面列出的基底材料的列表代表特定基底的实施例,但是所描述的转移技术应理解为基本上可与实质上任何基底一起工作。
从一个角度来看,可以提供一种化学气相沉积反应器,其被构造成使用前述方法中的任一种的方法来生产六方氮化硼。因此,可以提供一种化学气相沉积反应器,其可以生产获得上述效果和优点中的一种或多种的六方氮化硼。
从一个角度来看,可以提供一种控制器,该控制器被构造成控制使用前述方法中的任一种的方法来生产六方氮化硼的化学气相沉积反应器。因此,可以提供一种控制器,该控制器使得化学气相沉积反应器能够生产获得上述效果和优点中的一个或多个的六方氮化硼。
在审阅本公开之后,特别是在审阅附图简述、详述和权利要求部分时,其他方面也将变得显而易见。
附图说明
现在将参考附图仅以举例的方式描述本公开的实施例,其中:
图1:a示出了新型顺序种晶生长(ssg)h-bn生长过程的示意图。b描绘了在生长过程的各个阶段的基底的代表性扫描电子显微镜(sem)图像。
图2:a示意性地示出了在h-bn晶体的生长期间与成核和畴扩展有关的机制。b示意性地示出了在h-bn晶体的生长期间与均质化相关的机制。c示意性地示出了基底中活性物质的通量平衡。d示意性地示出了在生长过程中的不同示例点处相对于距基底表面的深度的活性物质的浓度。
图3:示出了气体类型在重结晶/种晶步骤期间对基底重结晶的影响。
图4:示出了温度对h-bn的成核速率的影响。
图5:示出了前体暴露持续时间在重结晶/种晶步骤期间对成核密度的影响。
图6:示出了持续时间在均质化步骤期间对解离度的影响。
图7:示出了在畴扩展步骤期间随着前体暴露持续时间的进一步成核程度。
图8:a示出了h-bn对基底的弱粘附性。b示出了用于生产层堆(layerstacks)(异质结构)的基于剥离的转移的过程流程图。c示出了使用转移方法产生的层堆的光学图像。
图9:示出了使用所公开的技术制造的示例设备的结果。
图10:示出了可以用于生长h-bn的类型的cvd室。
在附图中通过示例的方式示出了具体的示例方法,并且在本文中对其进行了详细描述,与此同时本公开易于进行各种修改和替代形式。然而,应当理解,所附的附图和详细描述并非旨在将本公开限制为所公开的特定形式,本公开将覆盖落入所要求保护的发明的精神和范围内的所有修改、等同形式和替代形式。
将认识到,本公开的上述实施例的特点可以以任何合适的组合方便地且可互换地使用。
具体实施方式
图1a显示了由本发明人发现的新型的h-bncvd生长过程的示意图,该过程被称为顺序种晶生长(ssg)。与常规方法一样,该过程分为退火阶段和生长阶段。然而,与常规方法不同,生长阶段本身分为三个不同的步骤。具体地,该新方法分为四个主要步骤:(a)退火步骤;(b)重结晶/种晶步骤;(c)均质化步骤;和(d)畴扩展步骤。
图1a用一组示例参数示意性地显示了这四个步骤。图1b显示了该过程内的示例时间点处的该组示例参数的sem图像。sem图像是从整个基底的各个位置获取的并且代表示例时间点的状态。所描绘的示例参数仅代表用于获得图1b所示的sem图像的特定组的条件。其他参数的示例在后面的附图中进行描述并在详述中进行相应的讨论。基于数值研究,我们估计除了下面将更详细讨论的明确测试的参数以外,其他参数将表现出创造性的效果。
退火步骤,即步骤(a),包括直至i点的时间段。退火步骤用于清除基底表面的碎屑,减少表面缺陷并引起基底一定程度的重结晶。在图1b中描绘的具体实施例中,铂箔被用作生长基底。使用的特定铂箔来自alfaaesar,厚度为25μm,且纯度为99.99%。然而,在sem图像i中所见,在步骤(a)后,铂箔仍然包含大量的单个畴;畴具有约0.1mm的典型畴尺寸。在实施例中,温度tgr升至1200℃且然后保持tan=15分钟。通过初步和数值研究,我们估计超过5分钟的任何时间都足以提供上述效果,但是明确考虑到可能在较短时间段处理的基底上生长h-bn。
重结晶/种晶步骤,即步骤(b),包括从i点到iii点的时间段(包括ii点)。从sem图像ii可以看出,在步骤(b)期间可以看到明显的进一步重结晶出现,使得铂箔看起来基本上是单晶的。在具体实施例中,将温度保持恒定,并将硼嗪在1x10-5毫巴的压力psd下引入cvd生长室长达3分钟的时间tsd。硼嗪的加入被理解为是促进这种显著进一步重结晶的关键因素。具体地,该机理被理解为是由在铂基底的硼嗪的催化分解引起的,在铂基底中释放的硼的一部分溶解在铂中。溶解的硼充当脱氧剂。它去除晶界处的抑制铂晶粒的生长的杂质。
在图像ii和图像iii之间没有明显的进一步重结晶。然而,图像iii确实显示了少数h-bn晶畴的初始成核。下面参考图2更详细地讨论形成它们的机制。相对于下面的图d、h和e,还探索了用于种晶/重结晶步骤的许多替代参数方案。
均质化步骤,即步骤(c),涵盖从iii点到iv点的时间段。在该步骤中,将硼嗪前体完全除去。在所示实施例中,温度再次保持恒定在1200℃。在图像iv中可以看到,在基底本身中没有看到进一步的变化,但是看到大的h-bn晶畴处于部分解离的状态,而小的h-bn晶畴已经完全解离。这在图像iv上的白色虚线区域内最为明显,其中可见对现有晶畴的损坏。该解离过程起到使在重结晶/种晶步骤期间形成的h-bn晶畴收缩的作用,使得仅在重结晶/种晶步骤期间形成的较大晶畴被iv点保留。在所描绘的实施例中,均质化步骤允许进行5分钟。将参照图2及其相应的讨论在下面更详细地讨论解离的机理。关于图6讨论了均质化步骤的持续时间的调整。
如上所述,通过本发明人确定的新型的ssg过程将晶畴的初始成核与这些畴的扩展分开。在ssg中,畴扩展主要在步骤(d)的畴扩展步骤期间发生。畴扩展步骤包括从iv点到vii点的时间段(包括v点和vi点)。调整该畴扩展步骤使h-bn晶畴的进一步成核最小化,同时提供允许畴扩展的条件。在所示实施例中,温度保持在1200℃下20分钟的时间段texp。值得注意的是,硼嗪前体的压力在2.5x10-6毫巴下显著低于重结晶/种晶步骤期间。这种较低的压力有助于抑制h-bn晶畴的进一步成核,同时仍然足以允许现有h-bn晶畴的扩展。
尽管在所描绘的实施例中,通过降低前体压力来相对于畴扩展抑制进一步成核,但实现该效果的其他参数方案也是可能的。成核过程和畴扩展过程均受温度、持续时间和压力的所有三个的影响。在进一步的附图和相应的描述中研究和讨论了各种替代方案。然而,从图像iv、v、vi和vii之间的晶畴尺寸差异可以看出,畴尺寸逐渐生长。具体地,进入畴扩展步骤(图像vi)的在15分钟的畴尺寸明显大于进入该步骤的在10分钟(图像v)的畴尺寸,这继而比该步骤开始时(图像iv)大得多。可以进一步看出,没有进一步的成核。这种缺少进一步成核将在下面结合图7进行进一步讨论。此外,通过图像vii,h-bn畴已经合并成连续膜。
现在转向图2,图2a和图2b示意性地示出了在h-bn晶畴的生长期间与成核、畴扩展和均质化相关的机制。图2c示意性示出了基底中活性物质的通量平衡且图2d示意性示出了在图1所示的生长过程中不同示例点处相对于距基底表面的深度的活性物质的浓度。
图2a和2b示意性地示出了在催化基底的表面上发生的各种过程。图2a显示了在ii点和iii点之间的重结晶/种晶步骤期间发生的四个主要传输过程。在暴露于前体(1)时,气体分子吸收到催化剂基底表面上并被催化离解。这些活性物种会解吸(2)或被吸收到本体(3)中。一旦催化剂表面的浓度超过饱和,成核就开始(例如,见核a和b)。通过活性物质在整个表面扩散并且将活性物质本身附着到边缘(4)核将生长。因此,基底中的浓度梯度对于成核速率以及该速率是正还是负是决定性的。一组类似的过程将出现在畴扩展步骤期间,但是具有较低的过饱和度,结果进一步的成核被抑制了。
图2b显示了在均质化步骤期间在iii点和iv点之间的传输过程。在该步骤期间,在没有其他前体以及高生长温度的情况下,现有核是不稳定的并开始解吸或扩散到本体(5)中。
活性物质的浓度梯度由活性物质的通量平衡决定。图2c中示意性地表示,在重结晶/种晶步骤期间,进入的通量jsd明显大于本体中的扩散通量jblk,导致基底表面出现较大的过饱和。相反,在畴扩展步骤期间,进入的通量仅略高于进入本体的扩散通量jblk,因此导致较小的过饱和。基底表面上的这种较小的饱和度足以允许现有h-bn晶畴扩展,但不足以允许显著的进一步成核。
为了清楚起见,针对两种方案已经显示相同的jblk,然而,jblk的值对每种方案不一定相同。例如,较高的温度将增加扩散速率,并因此增加从表面到基底本体的扩散通量。作为另一个实施例,基底本体中的活性物质的现有浓度越高,活性物质从本体向表面的扩散越大,因此从基底到本体的(净)通量越低。现有浓度将尤其取决于先前的前体压力和暴露持续时间生长室的温度的历史、催化基底的材料和催化基底的结晶度。
图2d示意性地显示了在如图1所示的生长过程中不同示例点的相对于距表面基底深度的活性物质的浓度。可以看出,在iii点,紧接重结晶/种晶步骤后,存在高水平的过饱和δcsd,这导致成核增加。因此,在将前体的分压保持在这些水平的同时,所得的前体暴露不仅导致现有核的生长,而且导致另外的成核。
相反,在iv、v、vi和vii点的畴扩展步骤期间,表现出小得多的过饱和δcgr。然而,这种饱和度会随着时间而累积,因此对于稳定施加的前体压力和生长室温度,浓度曲线变得更平坦并且另外的成核的可能性会随时间增加。这相对于在下面讨论的图7及其相应描述进行更详细的显示且用于在没有明显的进一步成核的情况下限制畴扩展步骤的有效最大持续时间。
图3示出了在重结晶/种晶步骤期间,气体类型选择对基底重结晶的影响。图3a总结了该比较测试的实验条件。具体地,将温度加热到1200℃,并在此温度下保持15分钟以进行图1所示的退火步骤。随后,保持真空,在10-3毫巴下加入氢气,在10-3毫巴下加入氨气或在10-5毫巴下加入硼嗪到生长室中,长达2分钟,作为“生长”步骤。
从图3b中可以看出,在真空、氢气和氨气条件下,仍保留相当多的结晶度。仅在存在含硼的气体的情况下,才能看到发生超出由退火步骤所引起的重结晶的进一步的重结晶。晶畴不同方向之间的亮度差异归因于通道对比度。
如上所述,这被理解为因为在铂基底的表面上通过硼嗪的催化分解而释放的硼溶解在铂中。硼用作脱氧剂,其可去除造成晶粒钉扎(grainpinning)的铂晶界上的污染物。根据数值模拟,我们了解到硼嗪可被如下替代:氨硼烷;三氯硼嗪;或氨气或氮气与硼酸三异丙酯、三苯硼烷、三氯化硼、乙硼烷或十硼烷的组合,因为这些化学物质在铂催化剂存在下以与硼嗪类似的方式催化分解。
图3c显示了购买的铂箔以及在使用上述条件在硼嗪中重结晶后的铂箔的x射线衍射(xrd)光谱。为了改善可见度,已经对光谱进行了补偿,其中后处理光谱乘以104。所有铂峰均标有米勒指数。标有*的两个峰来自安装铂箔的钽基座。可以看出,在处理后(111)指数占主导。应当注意,(222)指数等效于(111)。这表明实际上已经发生显著的重结晶。铂的(111)取向是热力学上最有利的状态,因为它是最紧密堆积的且因此需要最低的活化能。因此,预期在大多数条件下铂会优先以该取向结晶。
这种均匀的重结晶对于获得大面积的h-bn单晶畴非常有帮助,因为h-bn的取向本身受铂基底中畴的影响。应当理解,在h-bn晶畴的取向中存在适应于下面的基底晶格对称性或使晶格与基底失配最小化的竞争。在任一情况下,当h-bn晶畴穿过铂基底畴的边界时,由于h-bn晶畴试图匹配的优先取向的不连续变化,在h-bn中出现缺陷的可能性很高。因此,与其他可能的方式相比,通过硼的溶解的铂重结晶允许更大的h-bn晶畴。
图3d描绘了pt(111)反射(在2θ=39.7°)的纹理图,该纹理图显示了在对称位置(χ~0°)中的一个极点和在χ~70°和
图4示出了温度对h-bn晶畴成核速率的影响。图4a总结了用于该比较测试的实验条件。再次,首先将温度升高至预定水平并在该温度下保持15分钟以进行退火步骤。随后,在1x10-5毫巴下在与生长步骤相同的温度下引入硼嗪长达4分钟。然而,在该比较测试中,测试了三个不同的温度:1125℃,1200℃和1300℃。生长步骤结束后,立即淬灭样品并对具有h-bn晶畴的基底成像。
图4b显示了三个测试温度中每个的sem图像。注意到与其他两个图像相比,将1125℃图像上的比例放大,以提高可见度。可以看出,随着温度升高,整个系列的成核速率降低。这与上面关于图2的讨论所给出的作为cvd状态的基本生长模型的理论推论一致,即温度升高会导致更长的表面和本体扩散半径。
图5示出了重结晶/种晶步骤期间生长时间对成核密度的影响。图5a总结了该比较测试的实验条件。具体地,将温度加热至1200℃并在该温度下保持15分钟。随后,在1x10-5毫巴下引入硼嗪并在相同的温度下保持可变的分钟数,以进行生长步骤。在比较测试中,测试2、3、4、5和6分钟。生长步骤后,立即将样品淬灭并使用sem成像。
图5b显示了每个测试持续时间的sem图像。注意到与其他三个图像相比,2分钟和3分钟图像的比例被放大,以提高可见度。在2分钟下可见,对于选择的温度和压力,没有明显的成核。由白色虚线圆圈所示,只有在3分钟下才出现成核的开始。在超过3分钟的时刻,核继续生长,然而,在5分钟下,初始核被来自其他成核事件的另外的核连接。因此,尽管最终可能形成非常大的“岛”,但这些岛将由多个较小核的聚结形成且因此生长的h-bn将是多晶的,这对于高质量应用是不希望的。
上面关于图4所讨论的,除了温度对成核的影响外,前体气体的压力对成核也具有巨大的影响。这可以在图5中所示的比较测试(在1x10-5毫巴下进行)与仅在15分钟下表现出明显的成核的图7所示的比较测试(在2.5x10-6毫巴下进行)之间的对比中看出。下面将更详细地讨论图7。
图6示出了持续时间对均质化步骤期间解离度的影响。图6a总结了该比较测试的实验条件。将该比较测试再次分为退火阶段和生长阶段。然而,在该测试中,生长阶段被分解为生长步骤,然后是均质化步骤。整个温度保持在恒定的1200℃下。如前所述,退火步骤为15分钟。随后是生长步骤,其中在1x10-5毫巴下引入硼嗪3分钟或5分钟。然而,这时,在生长步骤之后是新型的均质化步骤。在此步骤中,除去硼嗪前体。测试了均质化步骤的三个不同持续时间,然后立即淬火并使用sem成像。具体地,分别测试了0分钟(即没有均质化步骤)、5分钟和10分钟。
图6b显示了对于三个均质化步骤持续时间在1x10-5毫巴下引入硼嗪3分钟时间的相应sem图像。在0分钟的第一图像中,我们看到两个描绘的h-bn晶畴都是完全完整的。这与其他两个图像形成鲜明的对比。在5分钟下,h-bn晶畴开始解离且损伤是可见的。在10分钟下,大多数h-bn晶畴已完全解离且其余畴的尺寸显著减小。这进一步支持了与以上图2的讨论有关的理论讨论。
图6c显示了对于三个均质化步骤持续时间在1x10-5毫巴下引入硼嗪5分钟时间的相应sem图像。在0分钟的第一图像中,我们看到存在两个不同尺寸的h-bn晶畴分布,所有这些分布都是完整的。同样,这与其他两个图像形成鲜明的对比。在5分钟下,大的h-bn晶畴开始解离且损伤是可见的,而小的h-bn晶畴消失。在10分钟下,大多数h-bn晶畴已完全解离且其余畴的尺寸显著减小。这再次进一步支持了与以上图2的讨论有关的理论讨论。
图7示出了在畴扩展步骤期间,随着前体暴露持续时间的进一步成核程度。图7a总结了该比较测试的实验条件。同样,将温度加热至1200℃并在该温度下保持15分钟。这之后是类似于关于图1讨论的畴扩展步骤,步骤(d),的低硼嗪压力生长步骤。具体地,在生长步骤期间在2.5x10-6毫巴下引入硼嗪可变持续时间。测试的三个持续时间分别为10分钟、15分钟和20分钟。同样,在生长后立即将样品淬灭并使用sem成像。
图7b显示了在三种情况下的相应的sem图像。注意到比例尺适用于所有三个图像。可以看出,在10分钟下没有成核现象。然而,到15分钟,观察到成核的开始。特别值得注意的是,此15分钟的开始时间大约是以上关于图5讨论的高压生长时间的五倍长。到20分钟,观察到更多的成核事件以及显著的畴扩展。
综合上述比较测试,可能会看到一些折衷,这些折衷使新型的ssg过程能够生长具有少量大面积h-bn晶畴的高质量h-bn。
作为一个实施例,与图5相比,在图7的较低前体压力条件下延长的成核时间允许一个时间段,在这种情况下长达约15分钟,此时预先存在的h-bn晶畴可以生长而没有明显的进一步成核的风险。这用常规的单生长步骤方法根本不可能。均质化步骤的存在进一步帮助了这一点,因为h-bn核的部分溶解确保了在重结晶/种晶步骤之后立即存在的小核很可能会完全解离,从而导致少量更均匀尺寸的h-bn晶畴进入畴扩展步骤。
如背景技术所述,开发新型的ssg过程的重要动机是生产具有少量大面积h-bn晶畴的高质量h-bn。然而,这种高质量的h-bn在不存在将h-bn转移到期望的器件基底而不损坏或污染h-bn的能力的情况下将具有有限的用途。此外,期望避免损坏催化基底,使得其可以重复用于多个生长循环。
如本发明人所认识的,能够转移的现有方法具有许多缺陷。一个特别的问题是在cvd生长目的中使用金属催化剂,该催化剂牢固地粘附到h-bn。强粘附性金属催化剂的实例包括镍、铁、钴、铑、钯和钌。因此,期望使用对h-bn具有弱粘附性的金属催化剂,例如铂或铜。
图8a示出了对基底的弱粘附性。所示的图像是在从cvd反应器中取出样品后5小时获取的h-bn在铂上的sem图像。可以清楚地看到弱粘附性,因为h-bn已开始自发地与基底解耦。为了清楚起见,突出了用外部虚线标记岛的外边缘且用内部虚线标记耦合(较暗)和解耦(较亮)区域之间的界限。
图8b示出了用于生产层堆(异质结构)的基于剥离的转移的过程流程图。在转移过程的步骤i之前,将合适的载体材料例如pva(聚乙烯醇)滴铸到已生长的h-bn上以形成“压模”。其他合适的载体材料包括lor(抗剥离剂)、pmma(聚甲基丙烯酸甲酯)、ppc(聚碳酸亚丙酯)、pvb(聚乙烯醇缩丁醛)、cab(醋酸纤维素丁酸酯)、pvp(聚乙烯吡咯烷酮)、pc(聚碳酸酯)或对h-bn的粘附性高于h-bn对生长基底的粘附性的任何其他合适的载体材料。
如果仅需要单层h-bn,则可以将pva压模简单地压在器件基底(例如二氧化硅)上,并且可以通过施加水溶解pva基底而不会损坏或污染h-bn。如果使用其他载体材料,则可以使用不破坏h-bn的任何合适技术来去除载体材料。在一些实施例中,这可包括使用合适的溶剂溶解载体材料,蒸发载体材料或将载体材料从h-bn中燃烧掉。还要注意的是,该剥落过程使生长基底不受损坏且可准备重新使用。
从一个角度来看,存在两种广泛的机制,可以通过所述两种广泛的机制去除载体层:溶解和破坏。可用于溶解载体材料的溶剂的实例包括丙酮、n-甲基-2-吡咯烷酮(nmp)、二甲基亚砜、己烷、苯、乙醚、二甲基甲酰胺、甲苯、乙酸丁酯、乙酸乙酯、乙醇、甲醇、氯仿、二氯甲烷、异丙醇和水。可用于破坏载体材料的技术实例包括等离子蚀刻(使用氢、氟或氧)、退火(使用氧或氢)和酸/碱处理(使用氢氧化钾(koh)、乙酸或硝酸)。
或者,如果需要层堆,则可以将pva压模与h-bn一起用于拾取其他层。在图8b的步骤ii、iii和iv中示意性地示出了该过程。可以以这种方式使用接合压模,因为h-bn层和在弱粘附性生长基底上的h-bn、石墨烯或其他二维材料的第二层之间的粘附性大于第二h-bn层、石墨烯层或其他二维材料层与其生长基底之间的粘附性。
图8c显示了使用该技术生产的示例层堆。左侧是二氧化硅基底上4层h-bn的光学图像。右侧是石墨烯层上方的h-bn层的光学图像,石墨烯本身在二氧化硅基底上。
图9a显示了经由使用上述技术生产的代表性h-bn/石墨烯场效应晶体管(fet)器件的4端子测量获得的传输性质。虚线显示仅偏移-0.2v的狄拉克点,代表了高质量石墨烯的固有性质。图9a的插图是使用所公开的技术制造的所测量的霍尔棒的光学图像。比例尺指示10μm。
图9b示出了霍尔迁移率测量。霍尔迁移率(μh)可以在不具有关于电容或曲线拟合的假设的情况下直接从该测量中提取。在室温下测量μh峰值=7200cm2v-1s-1。此外,测量n0=4.8x1010cm-2的低固有掺杂水平,确认低掺杂水平。在不需要任何高温退火以去除转移后的残留物的情况下实现这种低掺杂。因此,该示例器件用于证明可以使用所公开的技术来制造的高质量器件。
图10是示例性cvd反应器的示意图,其可用于使用新型ssg技术制造h-bn。将样品放在样品架中或样品架上。样品架可以是独立的元件或加热器本身的集成部分。通过使用一个或多个加热器来加热反应室。合适的加热器的实例包括电阻加热器、激光加热器或电磁加热器。在过程期间使用一个或多个温度测量设备监视温度。合适的温度测量设备的实例包括热电偶、高温计和电阻温度计。在过程期间使用一个或多个压力测量设备监视压力。合适的压力测量设备的实例包括电容压力表、皮拉尼压力表和电离压力表。前体气体通过一个或多个进气口进料到室中。通过控制一个或多个阀和泵来调节压力。合适的泵的实例包括膜泵、旋转泵和涡轮分子泵。可通过使用外部控制设备来控制反应器,该外部控制设备监控接收到的传感器数据并控制过程参数。
因此,将从以上描述显而易见的是,已经描述了一种用于生产大面积(与之前的几μm相比>0.5mm)的高质量h-bn的新型技术,其允许将生长的h-bn直接转移到所需的器件基底。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



